
Analogue Docking, Protein MD, Multiple Co-Folding Samples, Speed Estimates, and 2FA
docking analogues to a template; running MD on proteins w/o ligands; generating multiple structures with Boltz & Chai; runtime estimates & dispatch information; two-factor authentication; speedups




Today, we’re excited to announce two new workflows, an improvement to our co-folding workflow, a first pass at predicting how long workflows will take to run, the ability to enable two-factor authentication, and a few other speedups to Rowan workflows.
Analogue Docking
Rowan’s new analogue-docking workflow is useful in scenarios where you’re attempting to dock an entire series of compounds against a protein. The naïve way to do this is just to dock every compound against the protein. While this certainly works, it suffers from a few flaws:
Each molecule can find a different pose. Docking is a stochastic process, and it’s possible that different molecules might find different poses just by random chance, leading to apples-to-oranges comparisons. This can be ameliorated by using high exhaustiveness settings for docking, but at the cost of much slower workflows.
Imperfect alignment. Even when all the poses are “the same” (i.e. the conformers are qualitatively the same and the molecules overlay well), it’s not uncommon for there to be small alignment differences between analogues. This might not matter for routine docking screens, but it can cause issues for downstream free-energy calculations.
Wasted information. If you already know a bound pose and you’re docking closely related compounds, there’s typically little need to conduct an exhaustive search of all possible poses. Using the conformation of the input pose as a prior can lead to more efficient calculations.
Rowan’s analogue-docking workflow takes a protein, an input “template” pose, and a list of analogues (as SMILES strings). For each analogue, we:
Determine the maximum common substructure (MCS) for the analogue and the template.
Generate conformations for the analogue, subject to the constraint that the MCS must have the same coordinates as the template.
Check each generated conformer with PoseBusters to detect clashes with the protein.
For each valid conformer, run local optimization using the docking scoring function.
Finally, deduplicate the resulting conformations using PRISM Pruner.
For “close” analogues where only a few atoms are changing, this workflow typically returns poses with high structural overlap (≤0.15 Å RMSD).
The output poses can be viewed separately or overlaid with each other, as shown below. Hovering over each row shows the 2D structure of the molecule, and the table on the right shows the docking score, the RMSD to the template (in Å), and whether the pose passed the PoseBusters check or not.
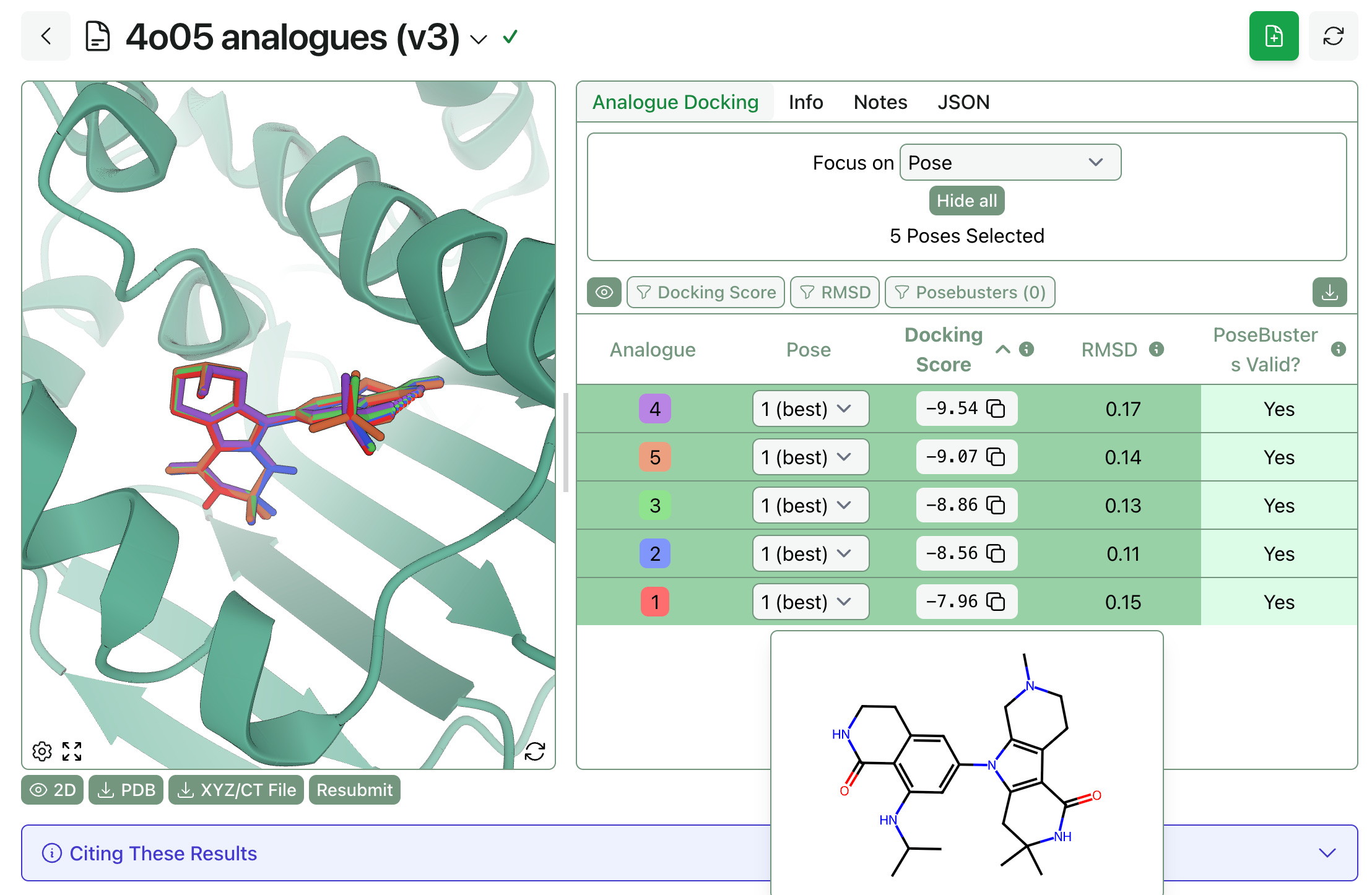
We anticipate that this workflow will be useful in hit-to-lead optimization scenarios—users can quickly enumerate analogues, submit them to analogue docking, and figure out which ones will be able to fit into the pocket. (While the different docking scores might occasionally be meaningful, we can’t promise that the best analogue will have the lowest score; relative binding free-energy perturbation would be the right way to answer that question.)
Protein-Only MD
Several months ago, we launched pose-analysis MD as a way to simulate protein–ligand complexes using molecular dynamics. Several users have requested a version of this workflow that doesn’t require a ligand, and today we’re launching “protein MD,” which works exactly like pose-analysis MD but without any ligand-related analytics.
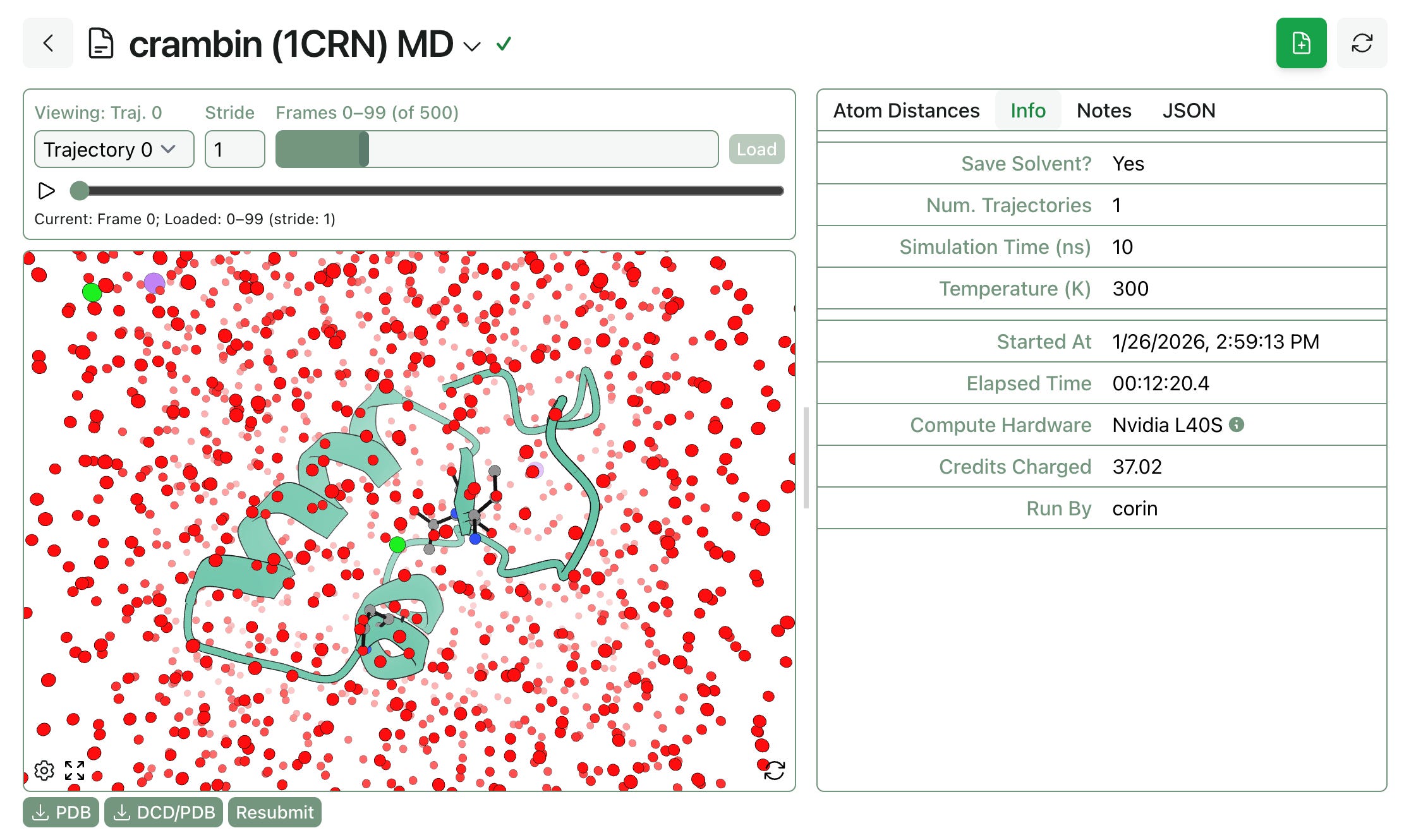
Protein MD is available to all Rowan subscribers, just like pose-analysis MD.
Multiple Samples for Co-Folding
Co-folding methods like Boltz-2 can be used to generate multiple protein–ligand poses; now you can tune how many poses Rowan generates when submitting a protein–ligand co-folding workflow. (Generating more poses takes slightly longer, but not as long as running an entirely separate workflow.)
To generate multiple poses, set a “Num. Samples” value greater than 1.
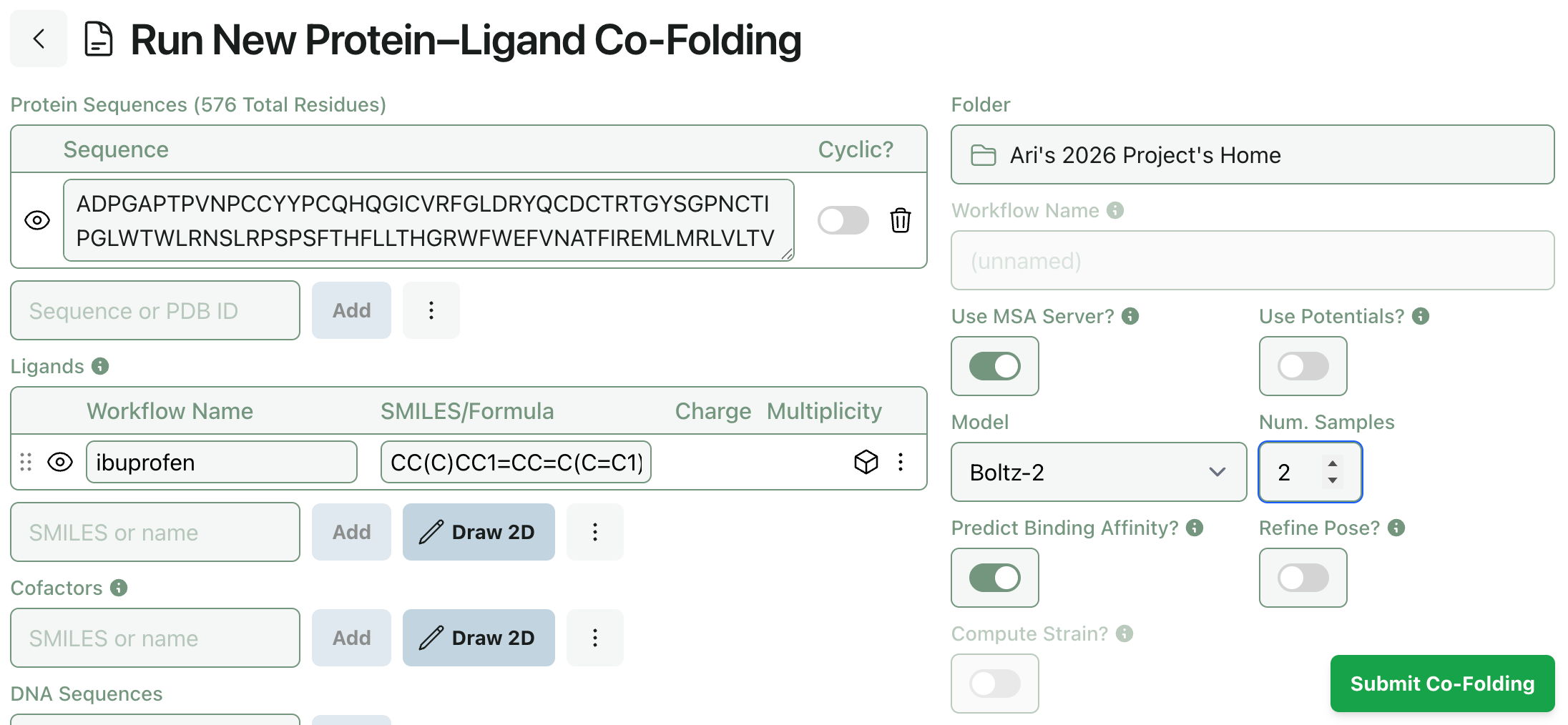
(When using Chai-1, this setting controls the model’s num_diffn_samples input. When using Boltz-2, this setting controls the model’s diffusion_samples input.)
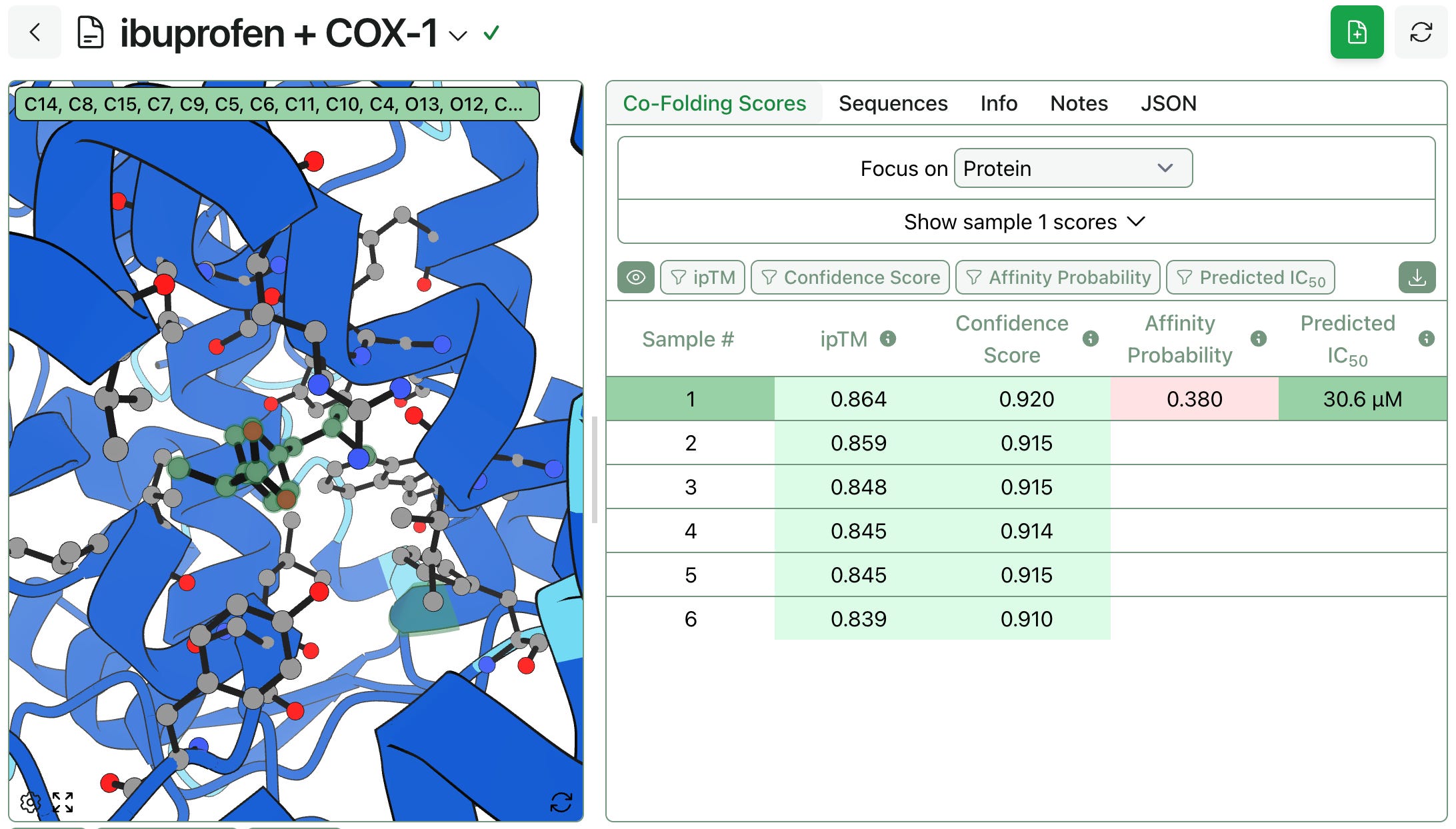
When using Boltz-2’s affinity prediction capability, the predicted binding affinity will take up to 5 samples into account. If more than 5 samples are generated, those with the highest ipTM scores are used. When storing and displaying this prediction, Rowan associates the affinity prediction with the highest ipTM sample.
Speed Estimation
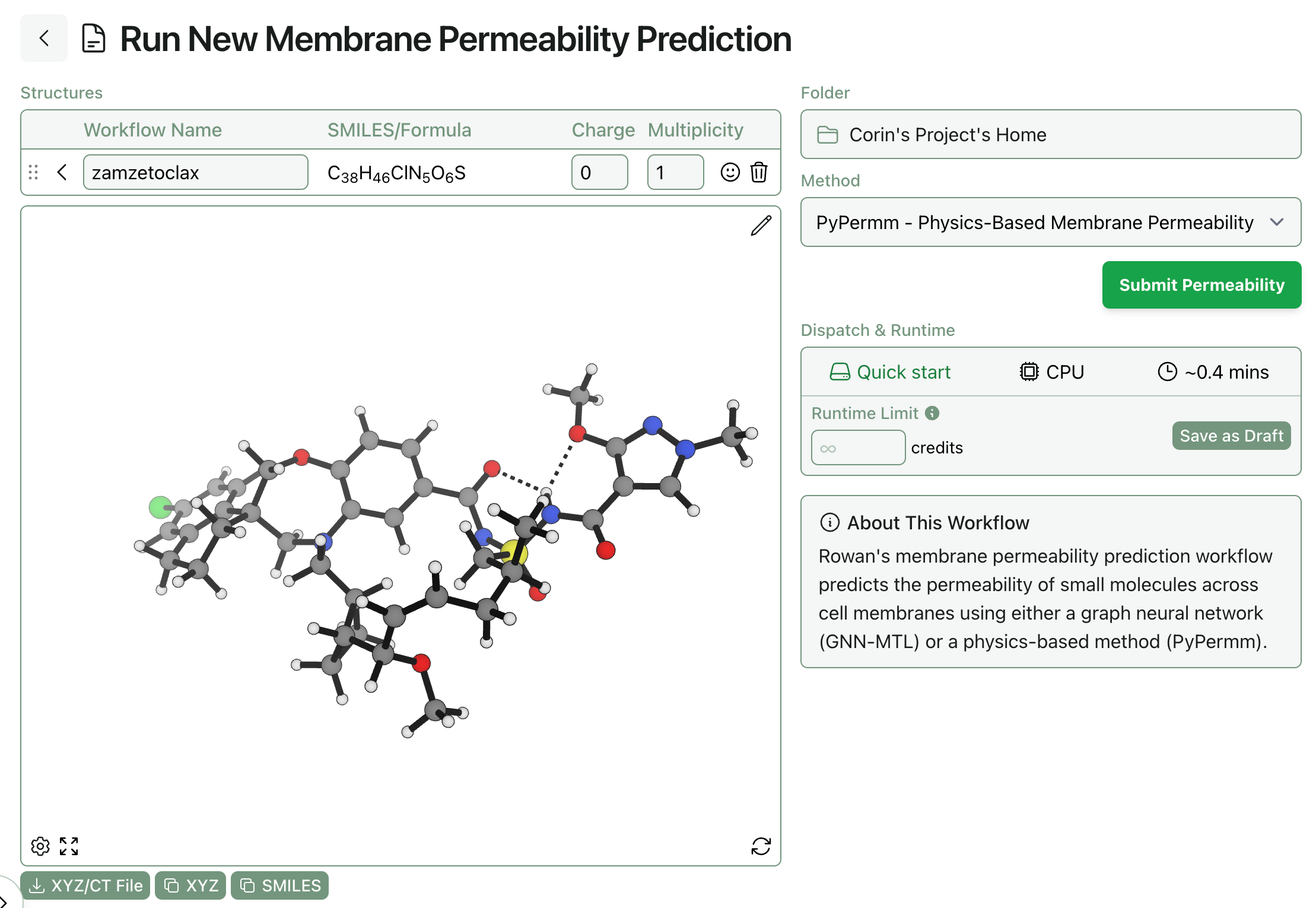
One of Rowan’s most requested features is the ability to predict how long a given job will take to run. Users want to know how long a calculation will take to run, which is very reasonable! To address this, we’re launching a new “Dispatch & Runtime” feature today. Before submitting a job, you can now see:
How long the workflow will take to start running. Some workflows run on hardware we always have allocated, while other workflows require us to allocate a whole new computer (which takes 3–5 minutes).
What hardware the job will run on. Some calculations go to CPUs, while others run on GPUs. Since the type of hardware dictates how many credits per minute the job will cost, it can be useful to know before submitting the job.
A rough estimate of how long the job will take to run. Some workflows have very predictable runtimes, and so we’ve been able to build a model of how long a calculation is expected to take. These are just rough estimates and are by no means guarantees—if you want to be sure a job won’t run for too long, set a runtime limit!
A brief note: Rowan takes the security and privacy of our users’ data very seriously, and we don’t train any models on user-computed data without being explicitly asked to. These runtime estimates are derived solely from workflows run by Rowan employees and not from any user data. (As always, users with security questions are welcome to refer to our terms of service or reach out directly to our team.)
The dispatch and runtime information can be viewed on the workflow submit screen, just below the “Submit” button:
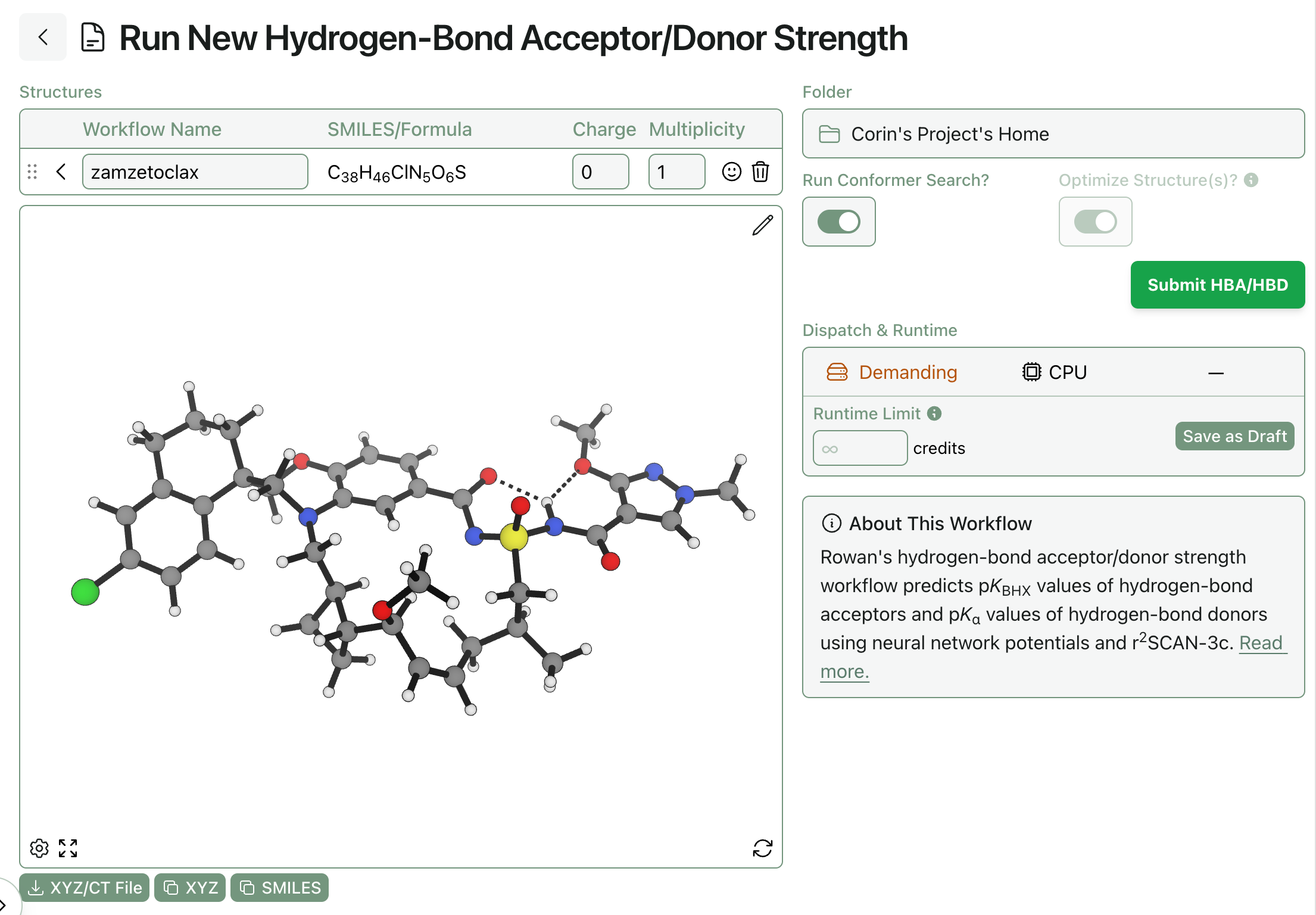
Because simulations involving electronic structure theory, geometry optimization, or conformational searches are highly chaotic and system-dependent, we’re not yet confident that we can provide runtime estimates with any useful level of accuracy for these calculations. Accordingly, certain types of workflows won’t populate that field, like the below hydrogen-bond-acceptor workflow:
Two-Factor Authentication
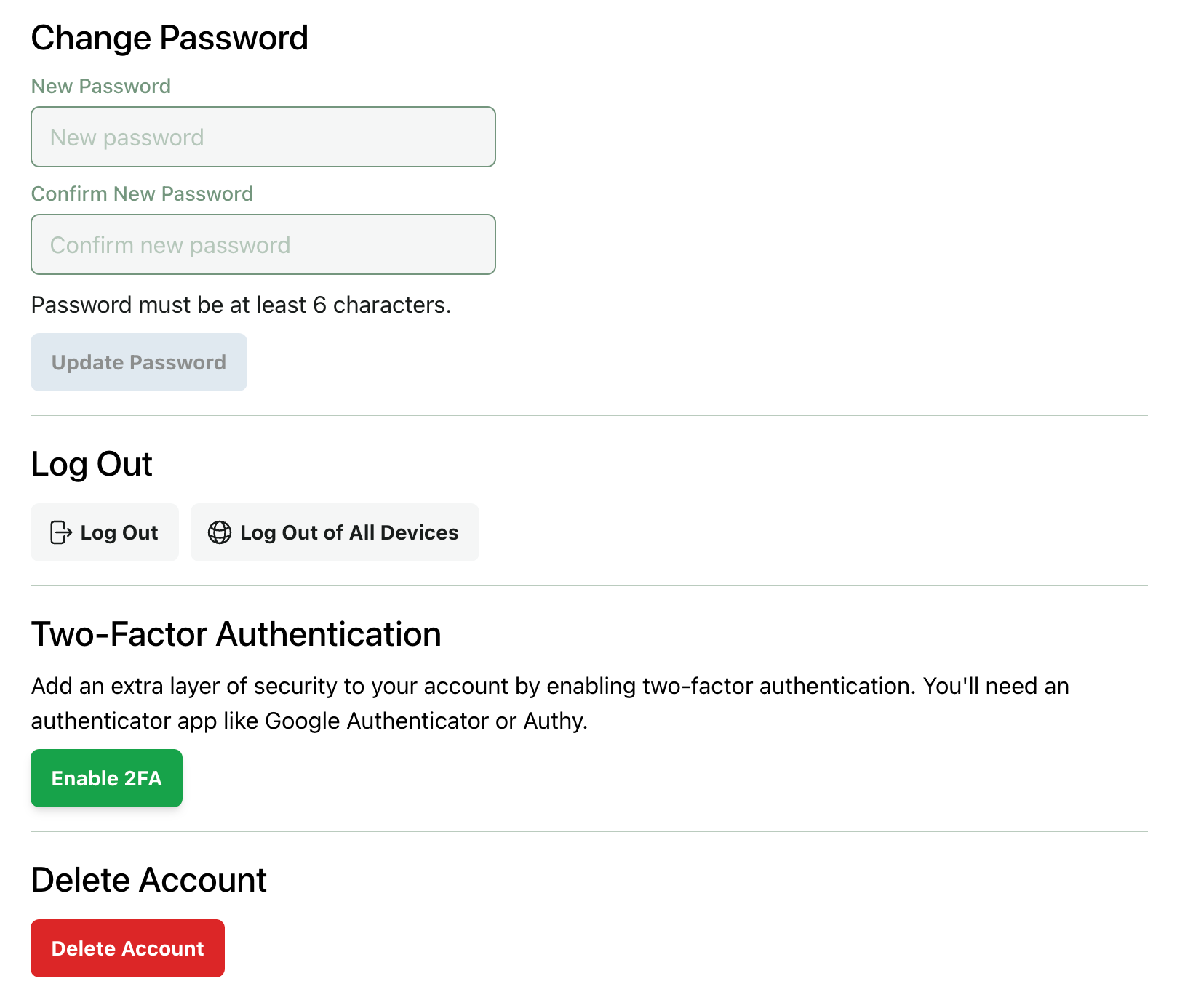
All Rowan users now have the option to enable two-factor authentication (2FA) as an added verification step when logging in. By default, 2FA is available but not required. If your organization would like to configure specific security requirements (ex. requiring all users to have 2FA enabled to access Rowan), our team would be happy to talk with you.
You can enable 2FA from your account page:
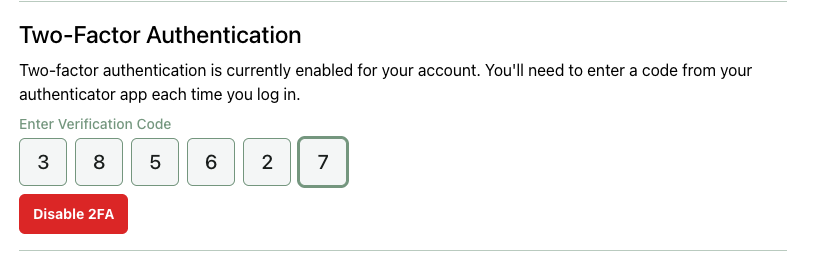
Once 2FA is enabled, you’ll need to complete a 2FA challenge after every login. 2FA can be disabled from your account page:
Various Speed Improvements
We’ve been working on a variety of speed and reliability improvements behind the scenes. While the exact algorithmic changes are a bit too prosaic to detail here, users should observe the following improvements:
Docking workflows that use conformer searches are noticeably faster (up to 2x).
Strain calculations run on poses generated by co-folding are similarly faster.
Macroscopic pKa calculations are more reliable for complex molecules.
As always, don’t hesitate to reach out if you find something that seems wrong. Happy computing!