
Co-Folding Updates
Boltz-2 FAQ and launch event recap; new visuals for co-folding workflows; new submission options; PDB bugfixes; new credit-management tools



Last Friday night, we posted an emergency Boltz-2 release. We’ve been thinking a lot about Boltz-2 over the past week. Our team attended the Boltz-2 launch event at MIT—Corin posted notes from the talk on X, which might be of interest for anyone who’s wasn’t able to attend.
We also put out a Boltz-2 FAQ which breaks down the paper and links to answers on social media about common Boltz-2 related questions, like “what GPU does this require,” “which binding-affinity output should I use,” and “how do these values stack up against experiment.” We’re updating this FAQ almost every day to keep up with the results pouring in from the community—if you’re not yet caught up, check it out!
Here’s what’s new on Rowan this week:
New Boltz-2 Visuals
Before we launched our first protein–ligand workflow, we built a protein viewer. This felt like a countercultural choice, as there are great open-source protein viewers like Mol*, but we wanted to be able to systematically improve our viewer over time. We’re still behind in some features, but we’re rapidly catching up.
This week, we added support for new ribbon color schemes: you can now color proteins by chain, residue index (rainbow), or pLDDT.
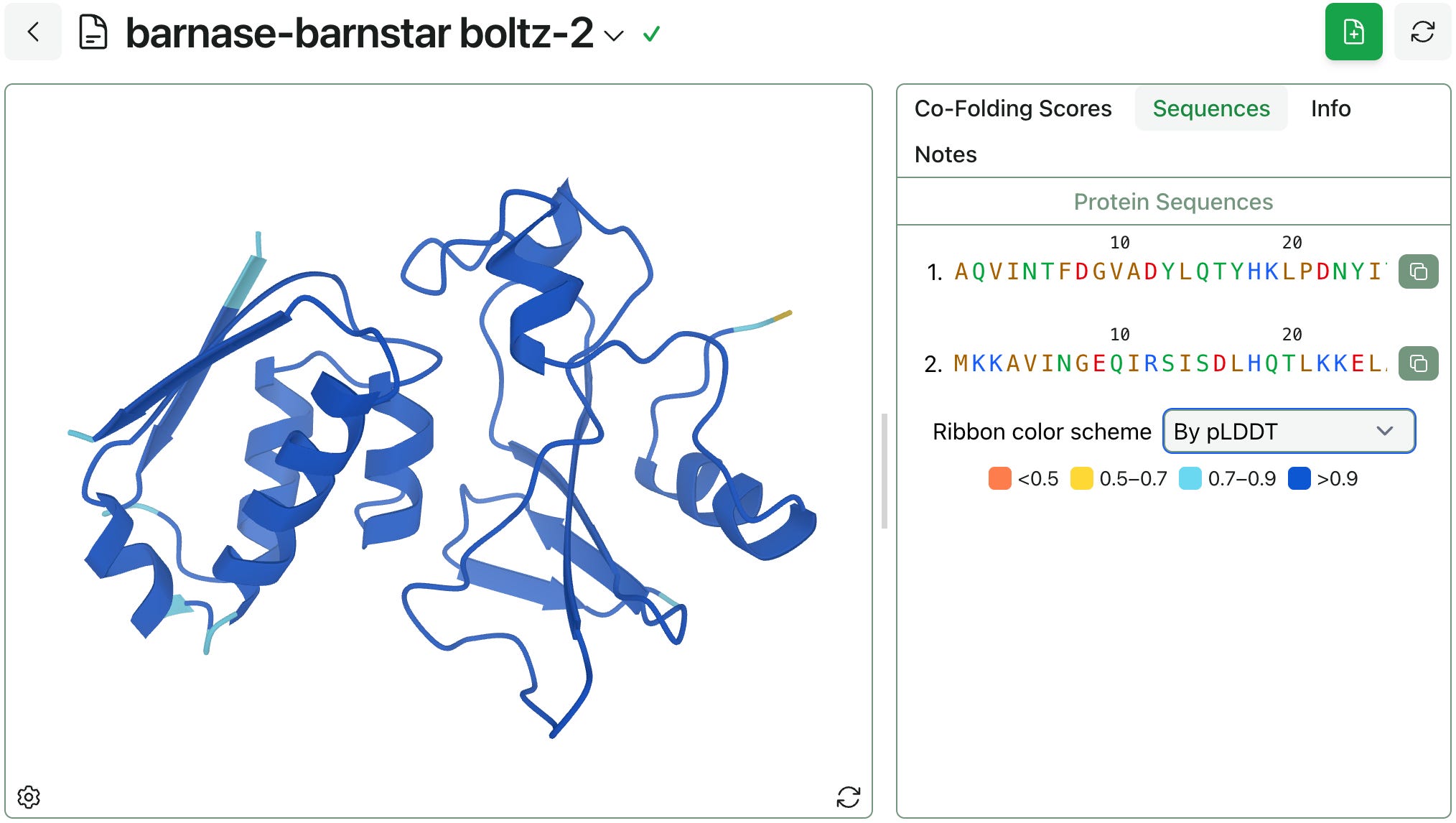
pLDDT is a common output of AlphaFold-style models. It’s a per-residue metric that predicts a structure’s Local Distance Difference Test (LDDT) values and indicates a model’s confidence in the local accuracy of a predicted structure. pLDDT ranges from 0 to 1 (higher is better); the above structure has a pLDDT of >0.9 for almost every residue, indicating high confidence in the predicted structure.
Having our own protein viewer lets us quickly make changes to respond to user feedback. If you have other suggestions for how we can improve our protein visualizations, please let us know at contact@rowansci.com!
New Boltz-2 Options
We’ve also exposed a “Use Potentials?” toggle for Boltz-1 and Boltz-2 that controls whether or not physics-based potentials are used to steer predictions at inference time. This helps improve the physicality of generated poses and preserve the chirality of ligands. (Turning “Use Potentials” on corresponds to Boltz-1x or Boltz-2x.)
After releasing Boltz-2, some users ran into issues with downloaded PDB files due to non-standard chain IDs like “A2.” We’ve updated our code to use single-character chain IDs and retroactively fixed existing protein records.
Credit Management
Scientific computing is powerful, but unpredictable. One of the most common questions we hear is “how long will this job take?” We don’t currently have much confidence in our ability to answer this question prospectively; structure optimizations using electronic structure methods involve iterative SCF cycles and geometry-optimization steps, both of which can vary wildly in runtime.
We’ve worked hard to give a general sense of job speed by warning when we think a workflow will take longer than 5 minutes—and we continuously track and improve the accuracy of those estimates.
But for longer jobs, “>5 minutes” isn’t specific enough. There's a big difference between 1 hour and 20 hours. To give you more control over credit usage, we’re introducing a new runtime limit setting
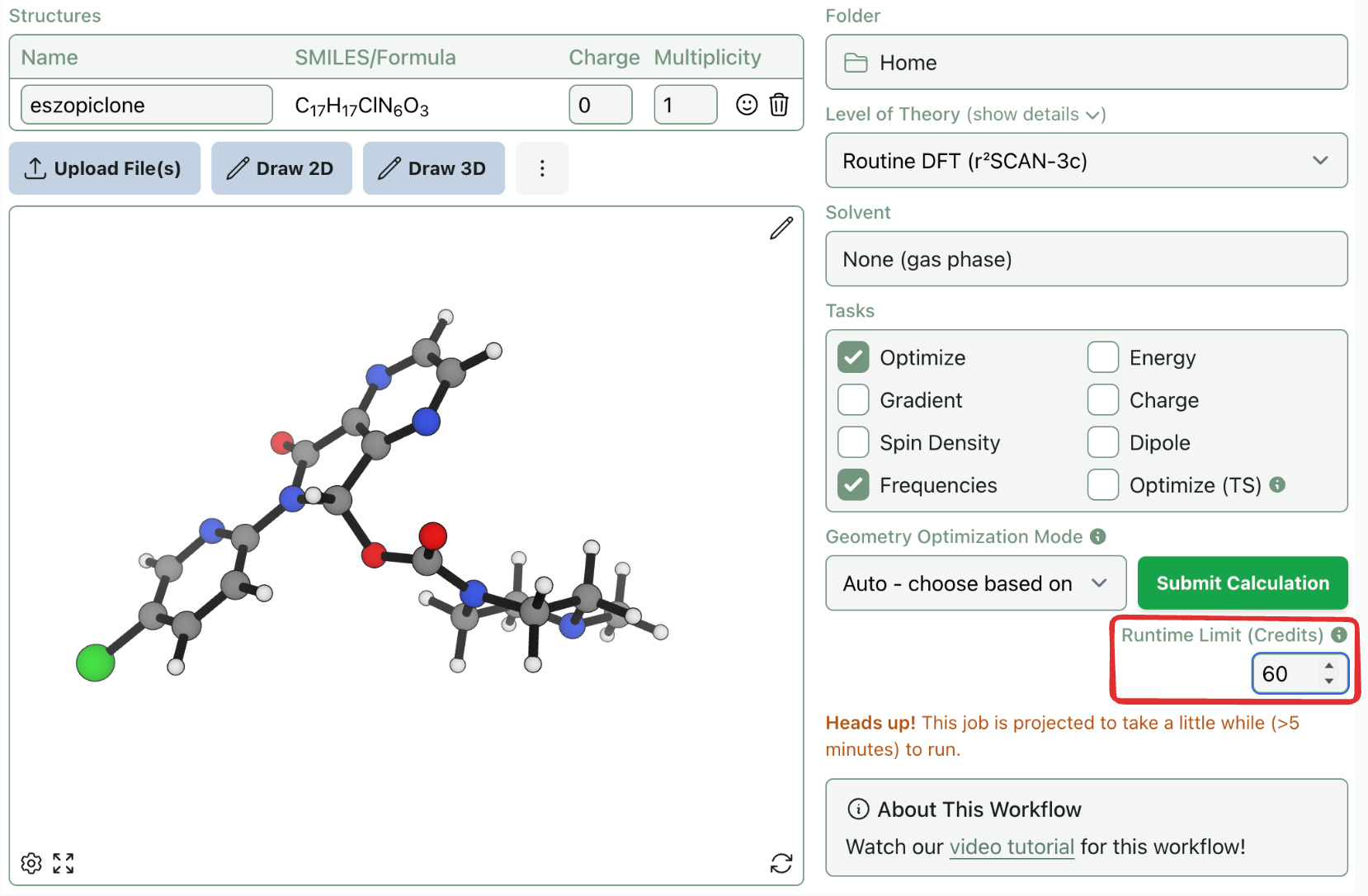
When submitting a workflow, you can now set a maximum runtime in credits (1 credit = 1 minute). If a job exceeds that limit, it will automatically stop. This limit can be updated or removed after a job is submitted. Due to the stateful nature of scientific software, jobs can't be resumed exactly—but optimizations can be resubmitted from the last saved structure, which is almost always lower in energy than your starting point.
We’re working on building more tools to give our users visibility into their credit usage on our platform. If you have suggestions for how we can improve this, please reach out!