
Conformer Deduplication, Clustering, and Analytics
deduplicating conformers with PRISM Pruner; Monte-Carlo-based conformer search; uploading conformer ensembles; clustering conformers to improve efficiency; better analytics on output ensembles




Conformational searching is an important task in computational chemistry, as almost every molecular modeling workflow requires accurate conformers. We’ve spent a lot of time working on Rowan’s conformer-search functionality: conformer searching was the second workflow we released, alongside tautomers and after pKa, and a year ago we released a comprehensive rewrite of the conformer-search workflow that added support for CREGEN deduplication and a variety of optimization methods.
Today, we’re excited to share a wide variety of updates that significantly increase the modularity, efficiency, and robustness of Rowan’s conformer-search workflow. These updates affect virtually every aspect of the workflow; the following infographic shows what happens during Rowan’s conformer-search workflow, with all the new additions shown in green.
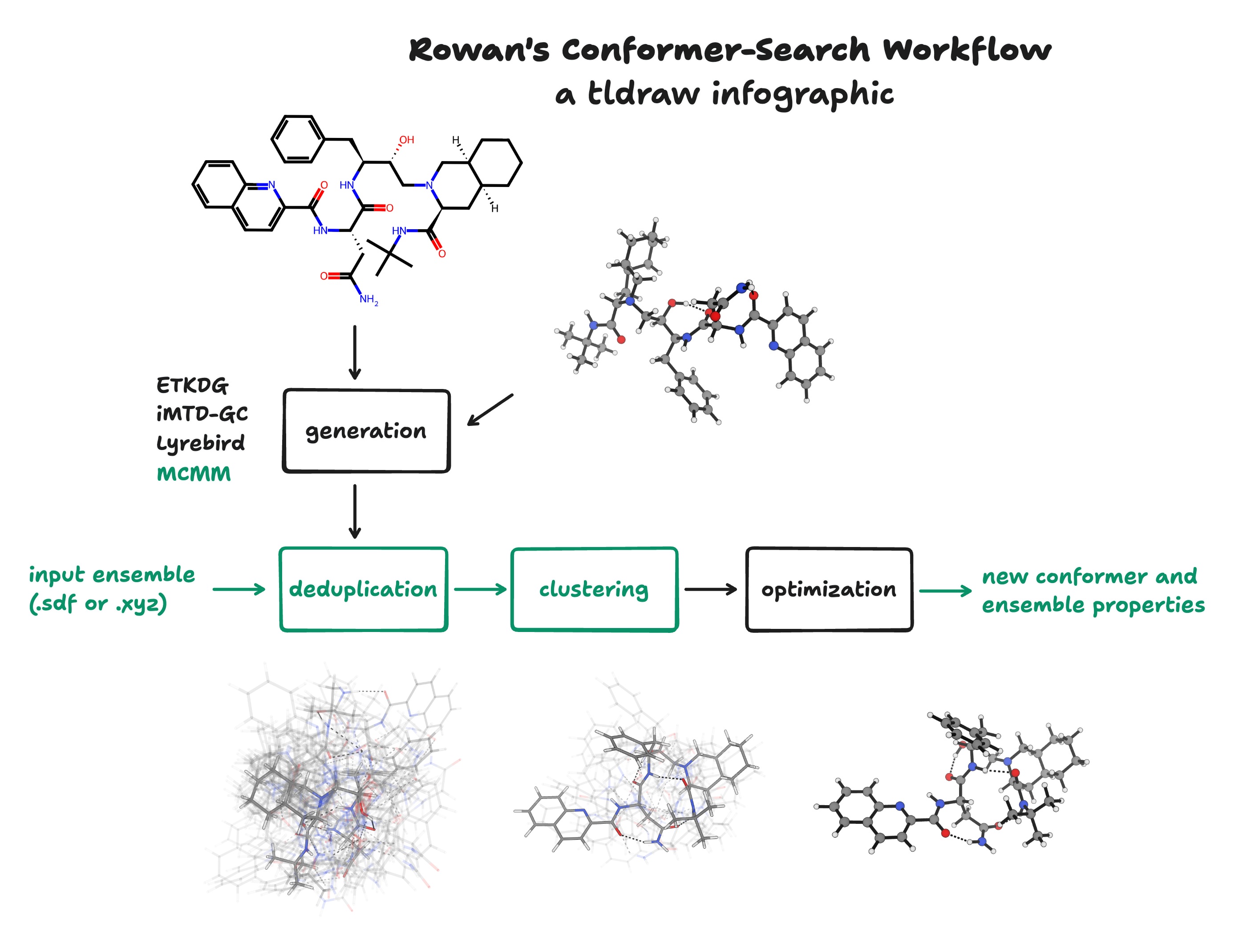
We wouldn’t have been able to do all of this without considerable help from (1) academic open-source partners at Yale and MIT and (2) our excellent pilot customers in industry who’ve helped us iterate and substantially improve our science. Thanks, everyone!
Conformer Deduplication
Many conformer generation methods stochastically generate numerous guess conformers that must be filtered down to a unique subset. RDKit’s ETKDG randomly generates torsional and distance parameters and then optimizes the generated conformers with MMFF94s. CREST’s iMTD-GC and iMTD-sMTD use metadynamics to explore conformational space and then optimize samples from the trajectory. During the process, these methods often produce a large number of duplicate conformers and rotamers, as well as structures that end up optimizing to the same structure. Additionally, further optimization of these ensembles with new methods can cause multiple generated conformers with different geometries to converge to the same structure.
These duplicates significantly increase the computational cost of downstream property prediction, so it is thus desirable to have a method of determining if two structures are truly distinct conformers. Crude optimization convergence parameters (used to minimize the number of optimization steps) can further complicate things by producing similar structures with slightly varying geometries.
Various methods are commonly used for conformer and rotamer deduplication, such as comparing the energies and moment-of-inertia tensors and atom-wise RMSD of positions. However, it can become prohibitive to perform an all-to-all comparison of conformers for large ensembles, since O(N2) comparisons are needed. RMSD can also hide local conformational changes in large structures as the contribution of a small local conformational change can be drowned out by the large number of atoms in the denominator.
There’s a need to efficiently prune the ensemble to a minimal subset without removing structures that are conformationally distinct. Nicolò Tampellini recently released PRISM Pruner, a python package dedicated to exactly this problem. He wrote a guest blog for Rowan about how it uses a a cached, iterative, divide-and-conquer approach to efficiently reduce the conformer ensemble to a unique subset. We now use this package to quickly prune conformer ensembles after conformer generation on the Rowan platform. The package is open-source, and if you have ideas of how to improve it, your contributions are welcome.
Multiple-Minimum Monte Carlo
As mentioned above, we’ve mostly relied on two conformer-search algorithms at Rowan:
The ETKDG algorithm (implemented in RDKit) uses a distance-geometry-based approach informed by experimental torsion-angle preferences to quickly build conformer ensembles. We often use ETKDG when we’re looking for quick and serviceable conformers for a downstream task, like when we’re building conformer ensembles just before docking.
The iMTD-GC algorithm (implemented in CREST) uses metadynamics and genetic crossing steps to explore conformational space. We typically use iMTD-GC when we’re looking for “slow and accurate” conformer search: for instance, in our ion-mobility mass spectrometry workflow.
(We also added support for the experimental Lyrebird flow-matching model recently: see the blog post.)
All of these methods can struggle when generating conformer ensembles for large molecules with lots of rotatable bonds: ETKDG often generates unphysical or very high-energy structures for complex molecules, while CREST’s metadynamics-based approach can struggle to explore large and complex conformational ensembles in reasonable amounts of time. We were thus very excited to see Nick Casetti (from the Coley Group at MIT) release an open-source implementation of Chang, Guida, and Still’s Monte Carlo conformer-search algorithm a few weeks ago.
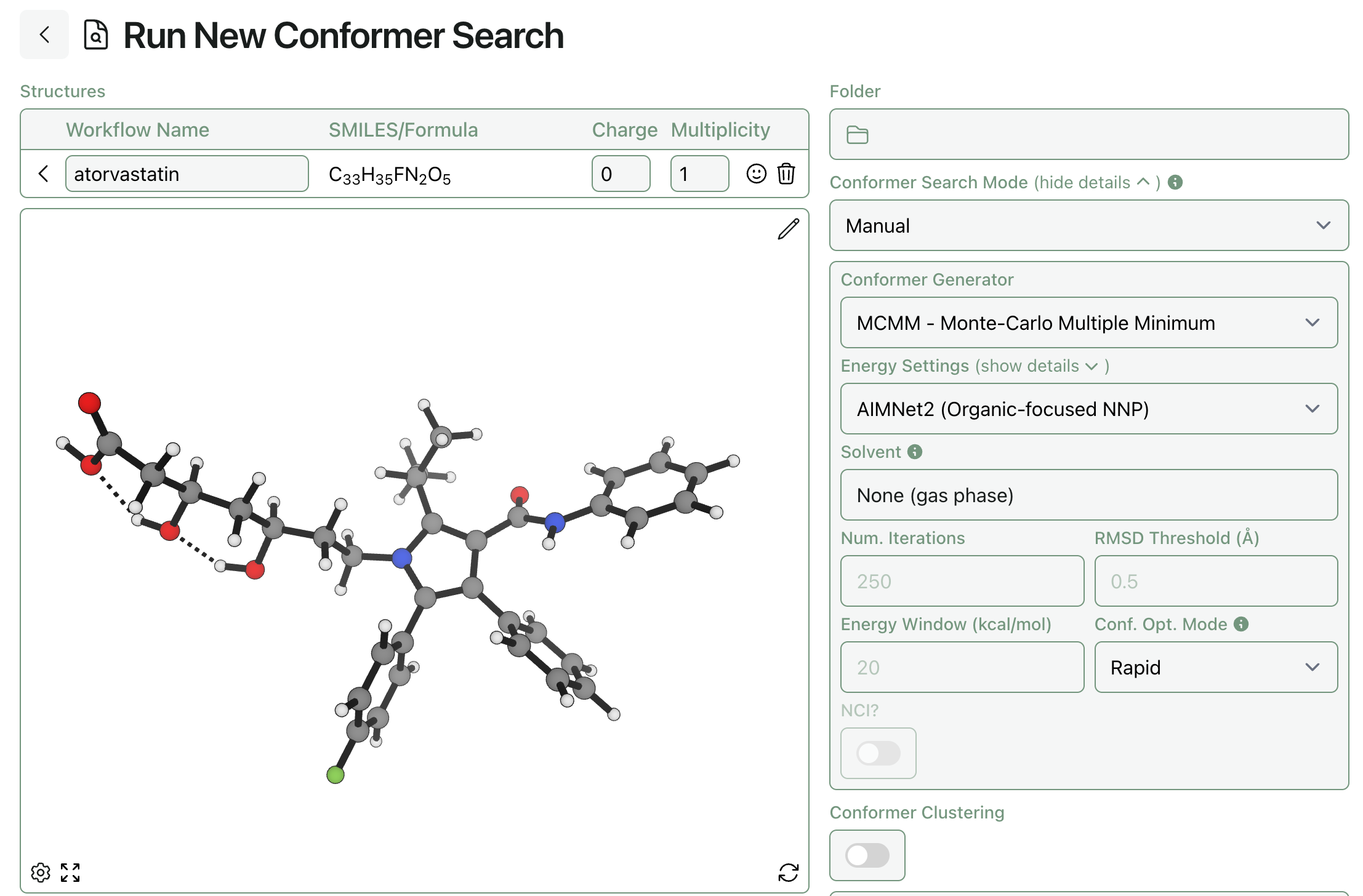
We’ve added support for Nick’s conformer-generation method (which we’re calling MCMM) in Rowan’s conformer-search workflow. Unlike CREST, which only works with xTB methods, you can use any level of theory with MCMM—and you can tune the number of samples, the energy window, and more through Rowan’s interface.
In our initial testing, MCMM performs similarly to iMTD-GC for small and medium-sized systems but starts to dramatically outperform iMTD-GC for very large molecules. For the “Naomi dimer” catalyst reported by Kennedy and co-workers, MCMM explores a much bigger region of conformer space than iMTD-GC while finding a significantly lower-energy structure:
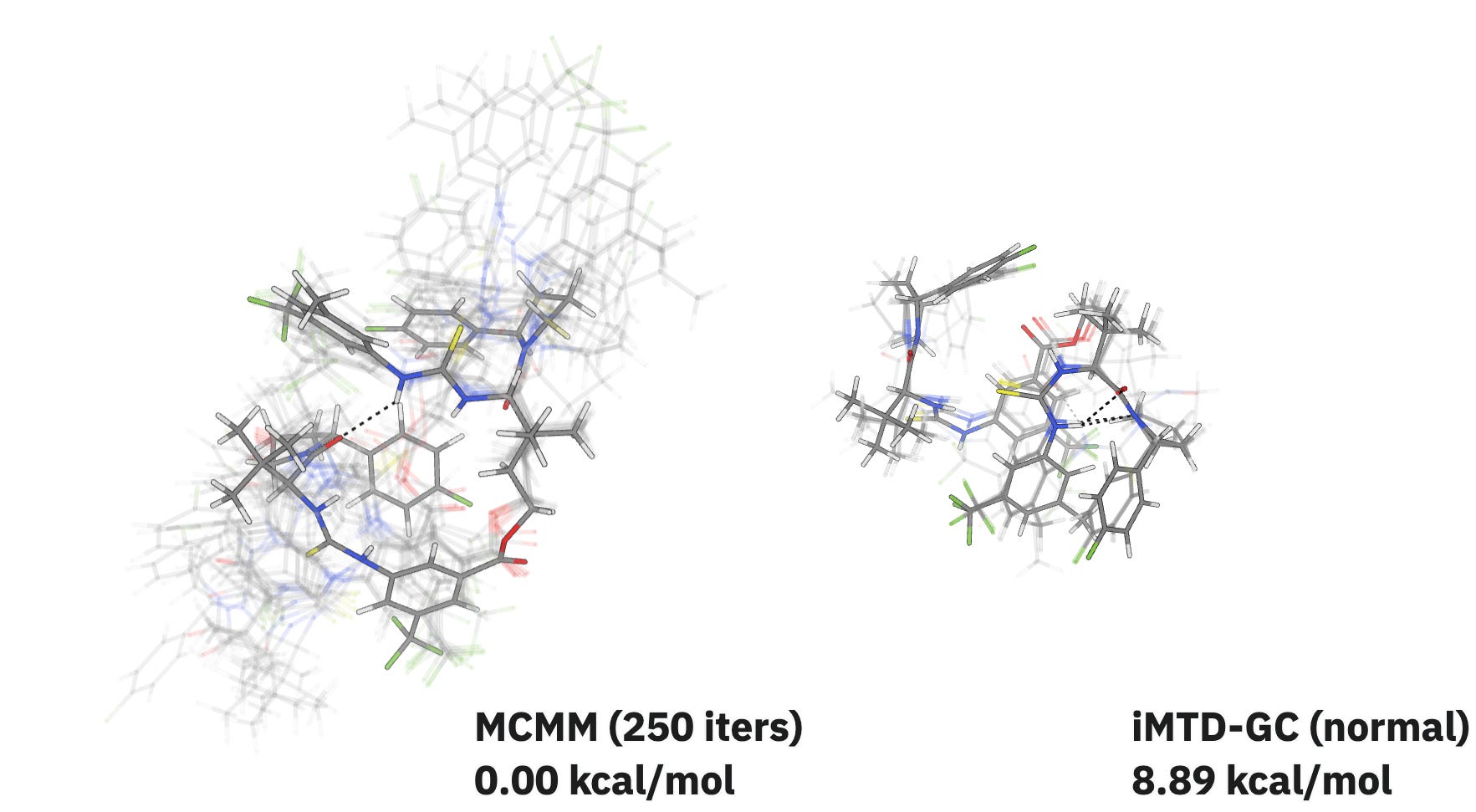
If you want to read more about how this method works and where it might outperform iMTD-GC or ETKDG, check out Nick’s guest post on Rowan’s blog!
Input Ensembles
While we’re working as hard as we can to make sure Rowan has the features that our users want, we realize that some of our scientists use many software packages—and that’s great! Rowan doesn’t (yet) have the best implementation of every algorithm, and we want to make Rowan easily interoperable with other scientific paradigms.
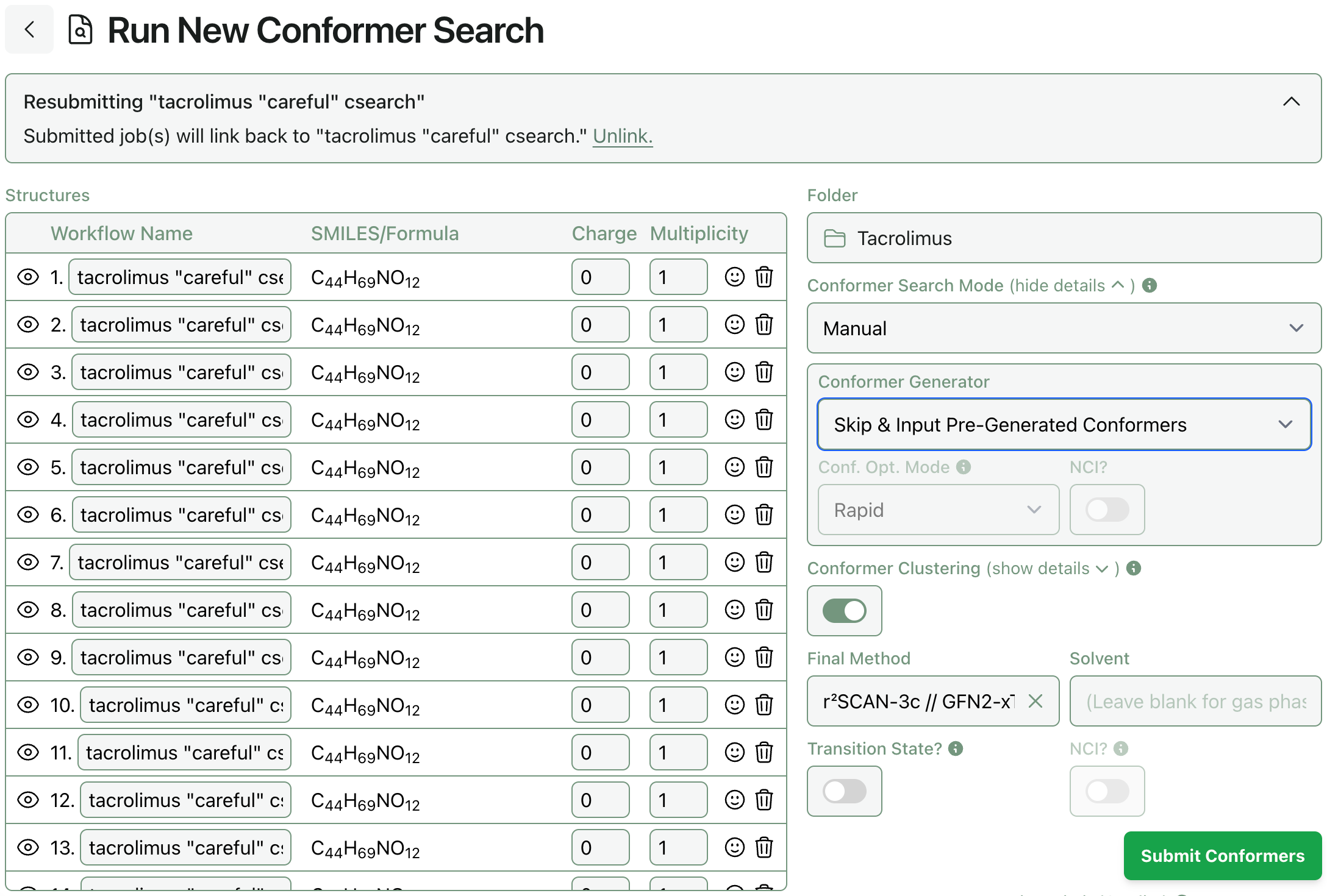
Some users have asked if they can upload conformer ensembles generated with other libraries, like Schrödinger’s MacroModel or OpenEye’s Omega, and run Rowan’s conformer optimization workflow on these ensembles to benefit from Rowan’s support for state-of-the-art methods (like NNPs and g-XTB) and convenient cloud infrastructure. As of today, it’s now possible to do this!
To submit an ensemble of pre-generated conformers to Rowan’s conformer-search workflow, select conformer-search mode “Manual” and set the conformer generator to “skip.” All the input structures will be submitted to a single conformer-search workflow that will start from the deduplication step.
Conformer Clustering
People run conformer searches for many reasons. For some applications, like spectroscopy, it’s essential to find the exact lowest-energy conformer; for other applications, it’s more important to sample diverse conformers in a cost-efficient way than to enumerate every potential rotational state of every group. To date, Rowan’s conformer-search methods have aimed at exhaustiveness rather than intentionally targeting diversity. This can be good in certain circumstances, but we’ve had users complain that methods like iMTD-GC return dozens of nearly identical conformers without sampling “worse” high-energy conformations that have substantially different shapes.
One of our users pointed us to ReSCoSS, a workflow developed by Anikó Udvarhelyi, Stephane Rodde, and Rainer Wilcken at Novartis. ReSCoSS solves the conformer diversity problem by performing an intermediate k-means clustering step before final conformer optimization; conformers are grouped into k clusters based on their overall properties, and then the lowest N conformers from each cluster are used to construct the final output ensemble. Udvarhelyi and co-workers show that ReSCoSS is a fast and effective method for computing ensemble properties like partition coefficients, prompting us to build our own ReSCoSS-like clustering step into Rowan’s conformer-search workflow.
Rowan’s conformer-clustering workflow uses a variety of three-dimensional descriptors to cluster conformers before selecting a user-tunable number of conformers per cluster. By default, Rowan groups conformers into 5 clusters and selects the lowest 3 conformers from each cluster, but this can easily be tuned. (Increasing the number of clusters ensures that more diverse conformers are obtained, while increasing the number of compounds per cluster increases the amount of conformers obtained for each region of conformer space.)
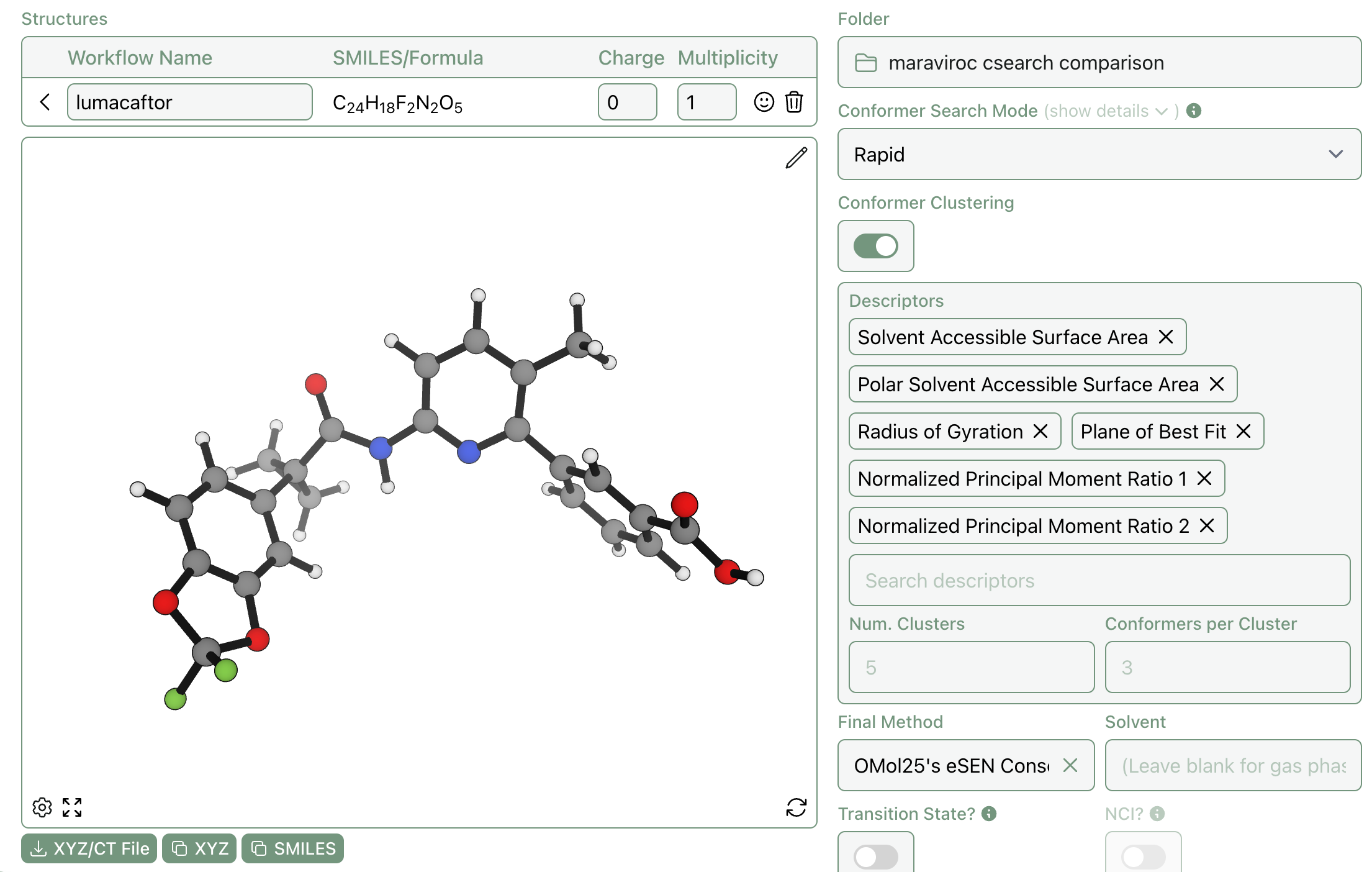
For large and flexible molecules, adding this clustering step significantly increases the speed of the workflow while still providing a diverse and representative set of output conformers. Conformer clustering is off in Rowan by default, but consider enabling it if you’re looking to generate diverse conformer ensembles in a cost-efficient manner!
Internal Coordinates
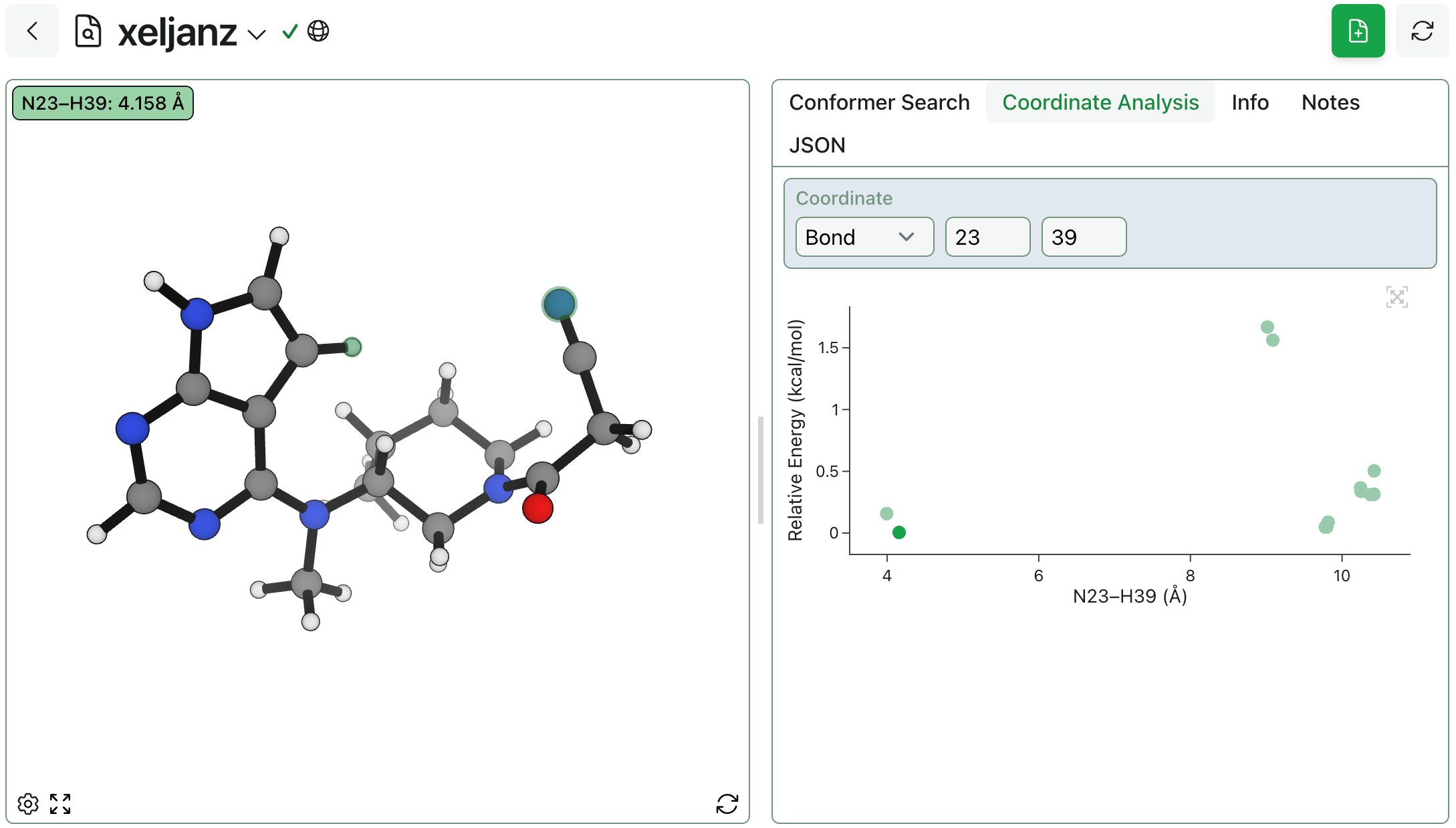
When dealing with ensembles of just a few conformers, it’s easy to glance at each structure (or compare the overlayed structures) and get a sense of which angles and torsions are relevant. However, this “visual inspection” approach doesn’t scale to large ensembles or flexible molecules.
To help with their analysis of conformer search results, we’ve had several Rowan users ask for the ability to select an angle and plot energy vs. angle, giving a more quantitative way of probing a structure’s conformational space. As of today, this is possible through the new “Coordinate Analysis” tab.
To plot relative energy vs. a bond, angle, or dihedral value, click into the “Coordinate” box and select atoms using the interactive 3D viewer (or enter atom indices). You can also click on the scatterplot’s points to change the view to the corresponding structure.
Conformer Analytics
We’ve also added simple output metrics to conformer-search workflows that help users understand how the overall shape and size of conformers differs. All conformer-search workflows now output certain properties for each conformer, as well as Boltzmann-averaged properties for the entire ensemble. To start, we’re reporting:
Solvent-accessible surface area, the surface area seen by a probe solvent molecule (in Å2).
Polar solvent-accessible surface area, the surface area corresponding to atoms other than carbon or hydrogen (also in Å2).
And radius of gyration, the root-mean-square distance of atomic mass from the center of mass (in Å).
We expect these properties to be useful in understanding how overall conformational ensembles change between different analogues or environments, as well as in quantifying differences between different conformations.
To show what this might look like in practice, we ran two conformer searches on a hydrogen-bonding molecular balance studied by Hubbard and co-workers. We can see that all three properties change substantially between various conformations:
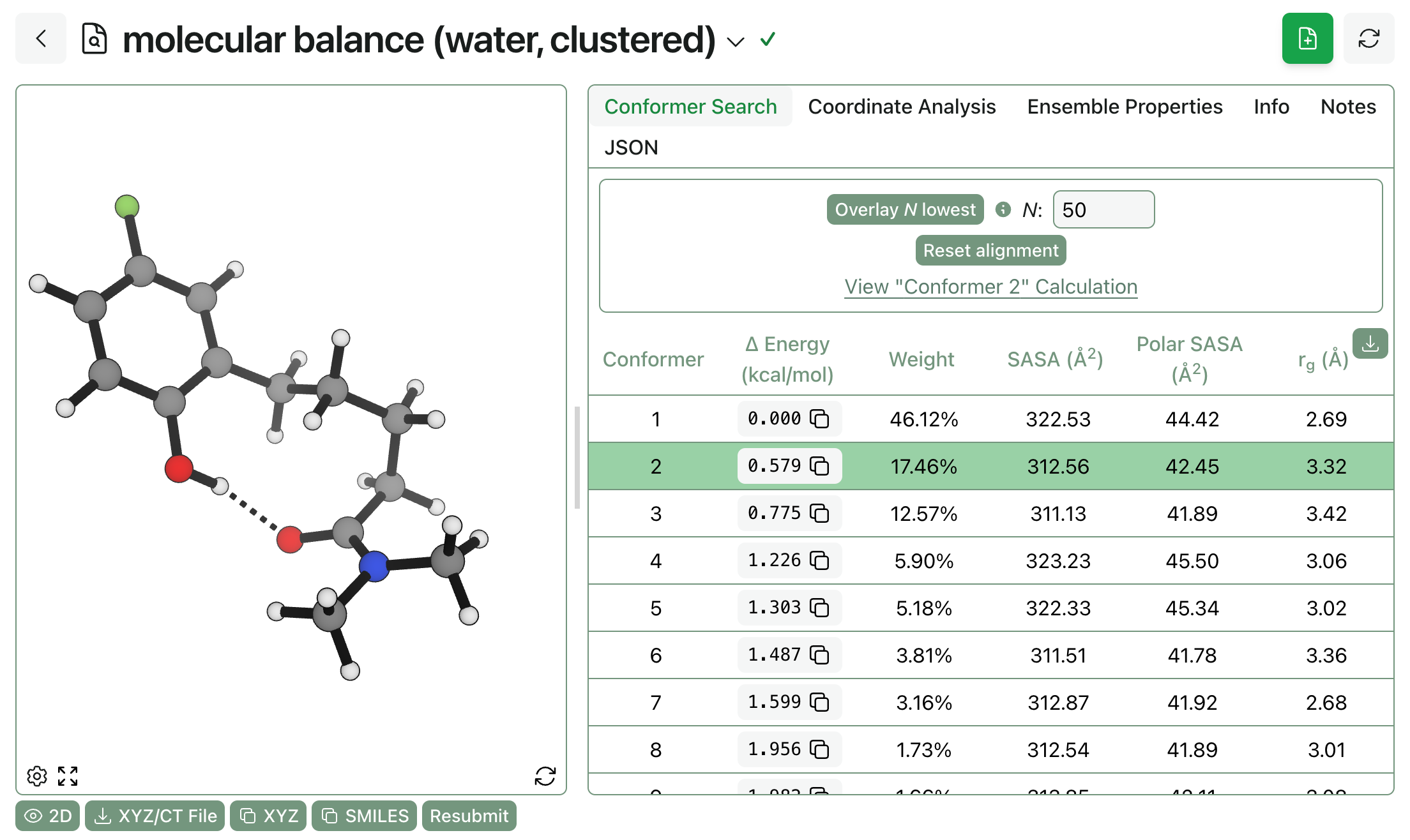
Furthermore, the overall ensemble properties change as the solvent changes: the molecule exposes more polar surface area in water than in hexanes but has a smaller radius of gyration because it doesn’t adopt the extended conformation with internal hydrogen bonding. (Here’s a link to the conformer search in hexanes and a link to the analogous search in water.)
We hope that these improvements make it easier to run faster, more powerful, and more accurate conformational searches in Rowan. Until next time, happy computing!
Love this!