
DiffLinker, MMFF94, and Common DFT Errors
iteration cycles in chemistry; generative AI for linker design; forcefield optimizations; handling common DFT errors


We’ve merged these emails with our existing user update emails, since our home-grown email update workflow has become cumbersome at scale. Users should have already received an email explaining this—but if you’re just reading this for the first time, welcome! We’re not trying to spam you: unsubscribed users have been omitted, and it’s now much easier to unsubscribe if you want. You may wish to read last week’s post on descriptors and PCA.

Programmers talk a lot about the “read–eval–print loop” (REPL), which allows programmers to write code, run it, and instantly view the output. These fast iteration cycles are part of what makes coding so dynamic: when you can try out ideas as fast as you can type, it’s possible to go to a hackathon and build an entire project overnight.
Chemistry, in contrast, is typically characterized by very slow iteration cycles. It takes weeks or months to synthesize a new molecule or material and try it out, which makes experimentation and design very slow. The promise of computational chemistry is that simulation can accelerate these design–make–test cycles, but historically even in silico work has taken a lot of time to run and analyze. Conventional workflows require the end user to write input files, transfer data to a high-performance computing cluster, queue and run the calculations (which can take days or weeks), transfer the results back, and visualize them in a separate software package—this whole process is often slower than running an experiment!
At Rowan, we’re building the infrastructure and software to make this whole process take seconds, not days. We’ve built a series of workflows that make it easy to set up complex computations, an integrated technology stack that runs complex calculations right away and a graphical user interface that gives publication-quality results even before a job has finished running. Now we’re taking the next step and launching two real-time features in our molecular viewer and editor, making it possible to iteratively design molecules without even having to queue a job.
Generate Linkers with DiffLinker
Today, we’re excited to release DiffLinker (paper, preprint, GitHub) to subscribed Rowan users. DiffLinker is a diffusion model trained to generate linkers to connect chemical fragments. DiffLinker generates linkers in two steps: first, a GNN is run to predict the size of a linker; then, an equivariant GNN is used to run the diffusion process of denoising the linker, resulting in a single linked molecule.

We think this work is really interesting for a few reasons! Instead of operating on string-based representations, it operates on 3D structures, allowing for more precise design of fragments that interact with a certain protein. This work is also the first of its kind that can link more than two fragments. Finally, it gives a good variety of responses and generates as many linkers as you ask it too—this can save you the time of digging through libraries of common motifs and looking for ideas. (Corin put together a blog post that goes into more detail here.)
This model isn’t perfect. Pat Walter’s blog post about it brings up several challenges:
Because DiffLinker only generates heavy atoms, assigning hydrogens and bond orders poses a challenge to integrating the model into workflows.
When generating lots of results for the same fragments, DiffLinker will frequently generate multiple conformers of the same molecule (in one test case, 145 of the 1000 generated compounds were of the same molecular graph).
Finally, DiffLinker doesn’t always generate stable or reasonably synthesizable results.
Even with these caveats, we think DiffLinker is a really interesting step in a promising direction, and we’re excited to integrate DiffLinker and other emerging generative AI technologies into Rowan.
Our interface to DiffLinker lives in Rowan’s molecule editor. To use DiffLinker, you can either draw fragments or upload them from somewhere else. In Rowan’s molecule editor, you can then click “Generate linkers with DiffLinker.” Within a few seconds, the model will return three linkers. We wrote a script to assign hydrogens to each of the generated linkers (though this is definitely not a perfect process yet), and you can use buttons or arrow keys to switch between the generated structures and either accept one or generate a new set of options.

We think that this sort of visual interface is one of the most intuitive and practical ways to interact with brand-new AI tools like DiffLinker. Users can always visualize the results of DiffLinker right away, making it easy to tell when DiffLinker gives unstable or bizarre results, and any useful ideas can immediately be simulated using our platform (e.g. with a geometry optimization). These tasks are much harder when you’re generating e.g. xyz files, where the presence of an O–O–O motif might not be immediately apparent.
We’re excited about finding useful ways to combine emerging generative AI tools and trusted simulation workflows. With this model now on our platform, you can start with promising fragments, generate as many linkers as you please, run lightning-fast optimizations on each of them with a neural network potential, and quickly evaluate the feasibility of each linked compound!
Clean Up Structures with MMFF94
Anyone who’s worked with 3D molecule editors before has run into this problem: you started with some structure, edited something small, and submitted an optimization, only for the structure to start flying apart or forming bonds you didn’t expect. Even when the molecule stays intact, it’s often the case that you spend 10–20 expensive DFT optimization steps fixing dihedral angles that a simple forcefield could have cleaned up right away. On account of this, a piece of feedback we’ve received a few times is that it’d be great to have a quick optimization method built into Rowan’s molecule editor to clean up a structure before submitting it.
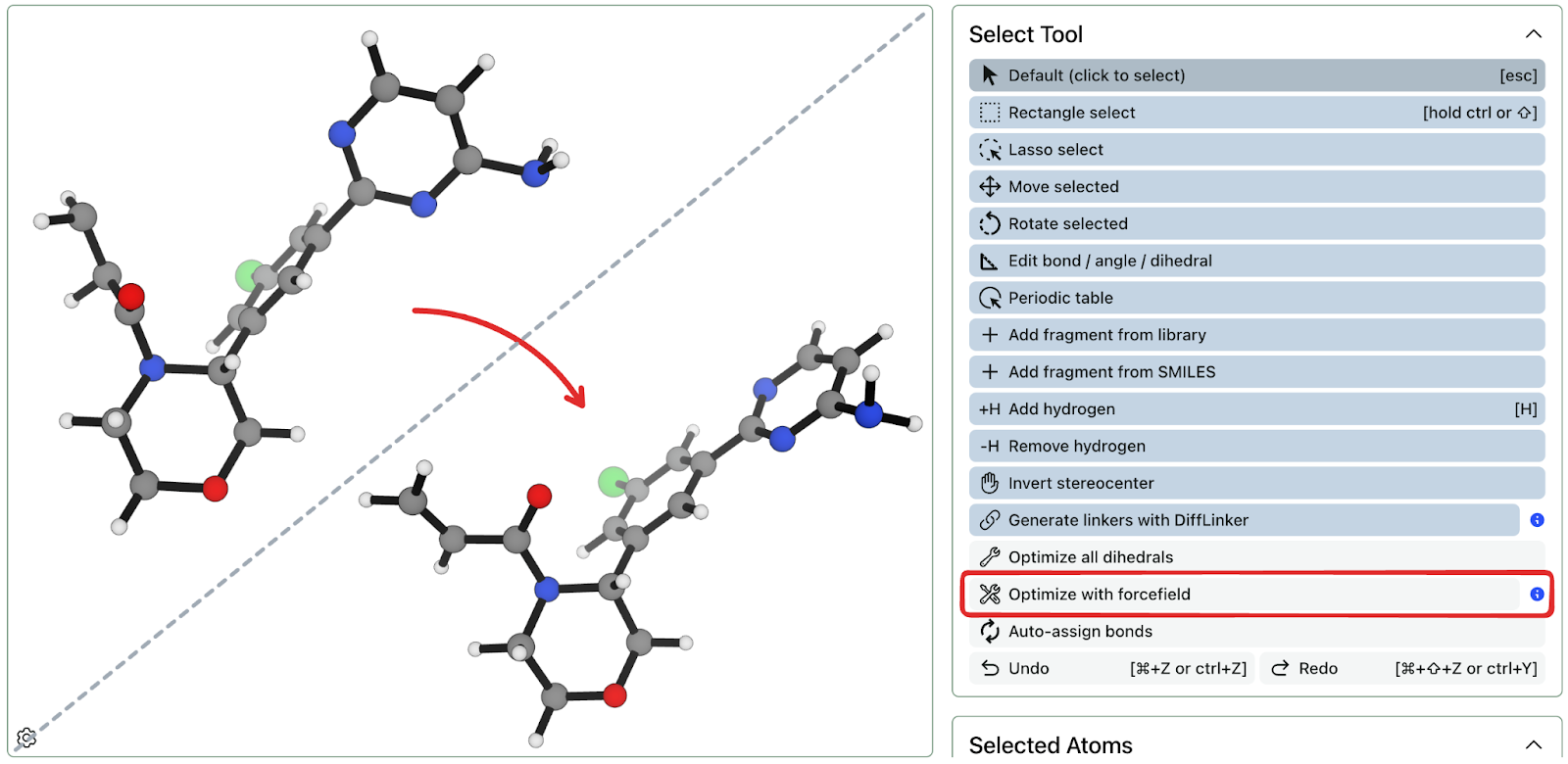
Today, we’ve added MMFF94 to Rowan’s structure editor. Subscribed users can use the new “Optimize with forcefield” tool, and Rowan will optimize the structure and return a result within seconds. Running these short forcefield optimizations doesn’t cost any credits—and it’s a great way to clean up bond lengths, angles, and dihedrals as well as confirming that your structure will behave like expected when you submit a workflow. (If you don’t like the optimization result, you can always hit “undo”.)
Because Rowan’s molecule builder is built for modifying XYZ structures, converting structures to a forcefield-parseable format can be bumpy right now—if you’re trying to run a MMFF94 optimization on your ionic crystal lattice, for instance, it will fail. In the future, we plan to extend this feature to work with more levels of theory and fix these problems, but we’ve already found this to be useful enough to ship. Let us know how it works!
Common DFT Errors
Density-functional theory (DFT) calculations are a cornerstone of modern computational chemistry, but there are a lot of ways that they can go wrong. Here at Rowan, we often see calculations that have been run with suboptimal or unreliable settings, which makes it difficult to have confidence in the output results. Read Corin’s full blog post on how Rowan handles DFT grid settings, symmetry handling, SCF convergence, and other common DFT errors.