
Double-Ended TS Search and the Invisible Work of Computer-Assisted Drug Design
finding transition states; the freezing-string method; using Rowan to find cool transition states; discussing drug design





Finding Transition States
Modeling reactions is either an art or a massive pain, depending on whom you ask. Even for simple reactions like a Diels–Alder cycloaddition, it can take a significant amount of time to generate the transition state (TS), optimize it, and confirm that it is the lowest energy pathway between the reactants and products. More complicated reactions involving large concerted motions can be even more painful: whole PhDs can be spent just looking for transition states, not to mention the large amounts of compute needed to run the DFT calculations.
The advent of neural network potentials has greatly accelerated the reaction modeling process. Transition state optimizations that used to take a day with DFT can now be completed in minutes. IRCs (which were often elided due to computational cost) can now be done in the time it takes to grab a cup of coffee. Conformational searches of transition states (formerly the domain of experts and limited to simple model systems) can now regularly be performed on 100-atom systems. The biggest barrier to automating reaction modeling is now often the initial generation of the transition state. Historically, many computational chemists opted for careful scans over a set of coordinates or templating from an existing transition-state geometry, but these approaches often take significant skill and chemical intuition.
Rowan is excited to be launching a new tool for finding transition states based on the freezing-string method, which greatly simplifies this process. The freezing-string method (FSM) starts from reactant and product geometries and forms “frozen” strings of geometries that expand towards the opposite string end while attempting to stay within the reaction channel. Once the ends are connected, the peak along the path is chosen as a guess for the transition state (TS) and optimized. (You can read more about double-ended-string methods in our Guessing Transition States blog.)
There are a lot of transition-state-finding methods out there—why use FSM? We've chosen to utilize the ML-FSM package developed by Jonah Marks and Joseph Gomes (ref, ref) because it provides significant improvements on existing growing string methods by using internal coordinate interpolation, L-BFGS-B optimization, and backtracking line search. ML-FSM is also a great example of industry–academia collaboration, with Jonah working closely with Jonathon Vandezande (our director of computational chemistry) to adapt the academic code into a modern Python package that can easily integrate with different methods. In our hands, FSM is incredibly robust to internal rotations, unoptimized starting geometries, and difficult transition states, and it's also very fast when paired with NNPs. We are planning to integrate other leading methods into our new double-ended TS-search workflow (and are excited about one day building a single-ended TS search).
Case Studies
Pericyclic reactions are often a particularly difficult class of reactions to model, with multiple σ and π bonds being simultaneously formed and broken in a cyclic transition state. These transition states are often nearly impossible to approximate with a simple scan, as multiple coordinates are involved in the reaction. The Claisen rearrangement is a simple [3,3]-sigmatropic condensation, forming a cyclic 6-membered transition state with multiple partially formed bonds. Using Rowan’s double-ended TS-search workflow, the TS can be found in two minutes with AIMNet2, with the workflow's only inputs being the reactants and products.

More complicated reactions can involve significant internal rotations or rearrangements of whole parts of the molecule. The divinylcyclopropane-cycloheptadiene rearrangement is driven by the release of ring strain, forming a seven-membered ring in the process. Rowan’s double-ended TS-search workflow finds the TS for this rearrangement in 5 minutes.
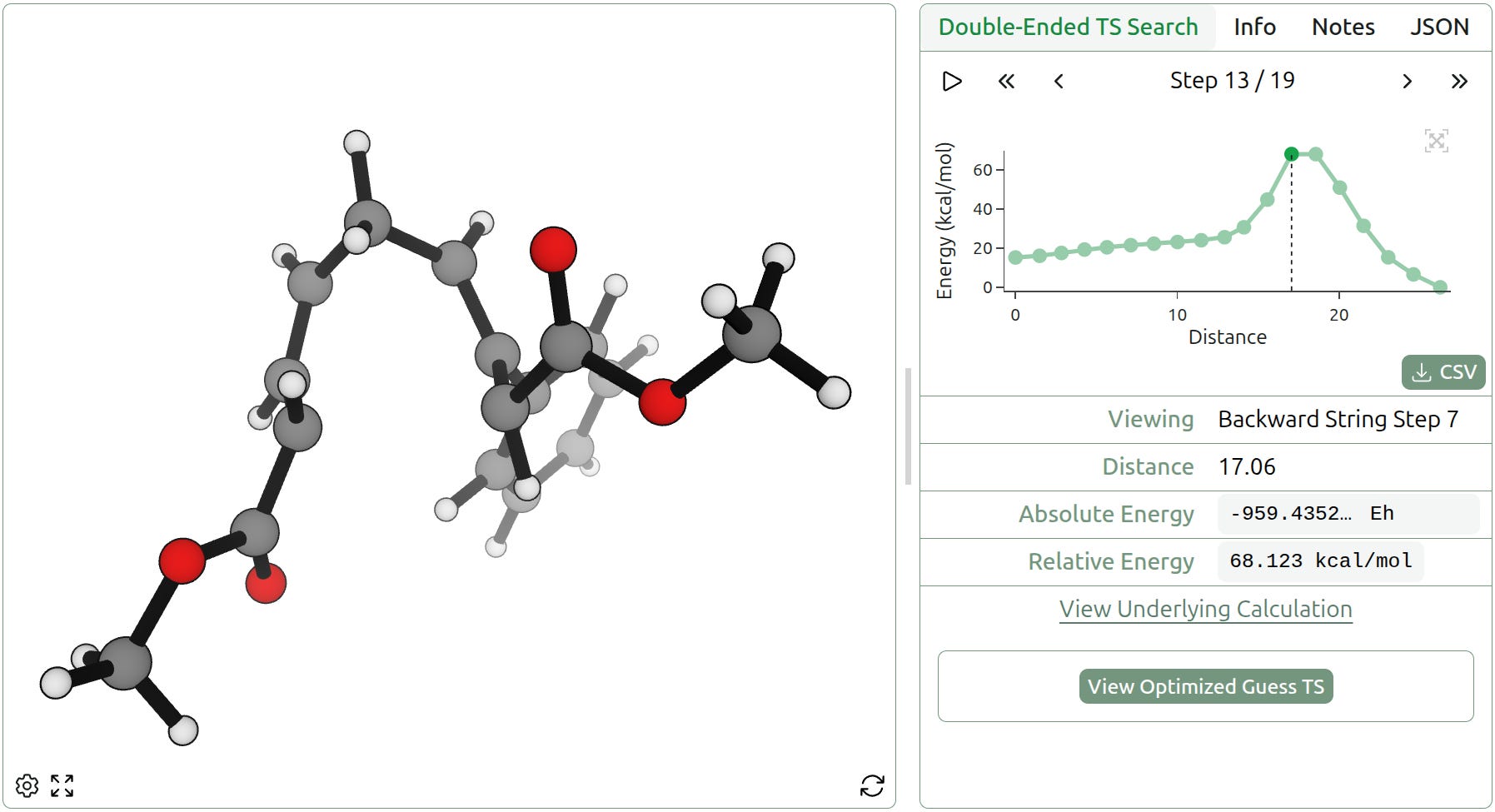
Double-ended TS search is even robust to intermediates. We examined a polycyclization reported in 2010 by Rob Knowles and Eric Jacobsen, which proceeds via elementary alkene-addition steps. Rowan’s double-ended TS-search workflow successfully identifies both constituent bond-forming steps and shows the singly cyclized species as a clear intermediate.

We stress-tested the FSM algorithm on the hydrocupration of 1-pentene, an unactivated terminal alkene, by BINAP•Cu(H). Alkene hydrocupration is an important and versatile way to functionalize alkenes, as the alkylcopper species generated can react with a variety of electrophiles; see this review by Richard Liu and Steve Buchwald. We anticipated that finding this TS might be challenging for FSM, as the BINAP•CuH species is bulky and contains many hindered rotors—however, we were able to get a reasonable guess for the TS in only 30 minutes. (We validated that the TS guess corresponds to the correct process with an IRC calculation.)

Rowan’s double-ended TS-search workflow joins a growing suite of reactivity modeling tools:
Transition states can be optimized through the “Basic Calculation” workflow.
The identity of these transition states can be confirmed by running IRC calculations.
A transition-state ensemble can be identified by running a transition-state conformer search.
The potential-energy surface along one or more coordinates can be studied using the “Scan” workflow.
Now, transition states can be located automatically using double-ended TS search methods.
All calculations can be run and analyzed through Rowan’s free Python API, making it simple to run hundreds of GPU jobs at once or quickly screen different levels of theory. This can be hugely enabling: in the course of an eight-week internship, our intern Sawyer has run over 14,000 workflows using Rowan’s API.
We think that this suite of tools is ideally suited to automating reactivity modeling and enabling high-throughput catalyst optimization or mechanistic study. If you’re interested in exploring how to migrate your reactivity modeling tasks to Rowan’s modern platform, please reach out to our team at contact@rowansci.com—we’d love to talk.
The Invisible Work of Computer-Assisted Drug Design
The workload for computer-assisted drug discovery (CADD) scientists is not getting any lighter as CADD software gets more sophisticated and complex. Building a state-of-the-art CADD apparatus requires a lot: evaluating software solutions, benchmarking them, testing on internal data, integrating tools into a larger ecosystem, and maintaining robust data interconversions. And all of this is only half the battle; armed with this machinery, CADD teams need to integrate computation into the experimental design–test–analyze cycle to provide value.
In a recent blog post, we look at the “invisible work” of CADD teams that’s required before they can be impactful. At Rowan, we’re working to solve these problems by building a platform that gives CADD teams a head start. Read the full blog post here.
Discounts for Annual Billing
You can now subscribe to Rowan annually and enjoy discounted rates. Save 15% (that’s $360/year) on industry plans or 10% on academic plans with annual billing. You can subscribe to the annual plan through the Rowan platform. (Or, if you’re already subscribed with monthly billing, you can email us at contact@rowansci.com to switch.)