
Free-Energy Perturbation
what FEP is and why it's useful; limitations of current methods; Rowan FEP, TMD, and public benchmarks; how to run FEP in Rowan; the dream of FEP "too cheap to meter"; how to try Rowan FEP




Today, we’re releasing our biggest feature to date: free-energy perturbation (FEP), a physics-based method for predicting protein–ligand binding affinity.
In this post, we’ll briefly give some scientific context for what FEP is and why it matters, discuss Rowan’s approach to FEP and its advantages, and explain how interested companies can use Rowan FEP. (Experienced drug-discovery readers may already know exactly what FEP is and why it’s useful; if this describes you, feel free to skip the first section of this post.)
What is Free-Energy Perturbation?
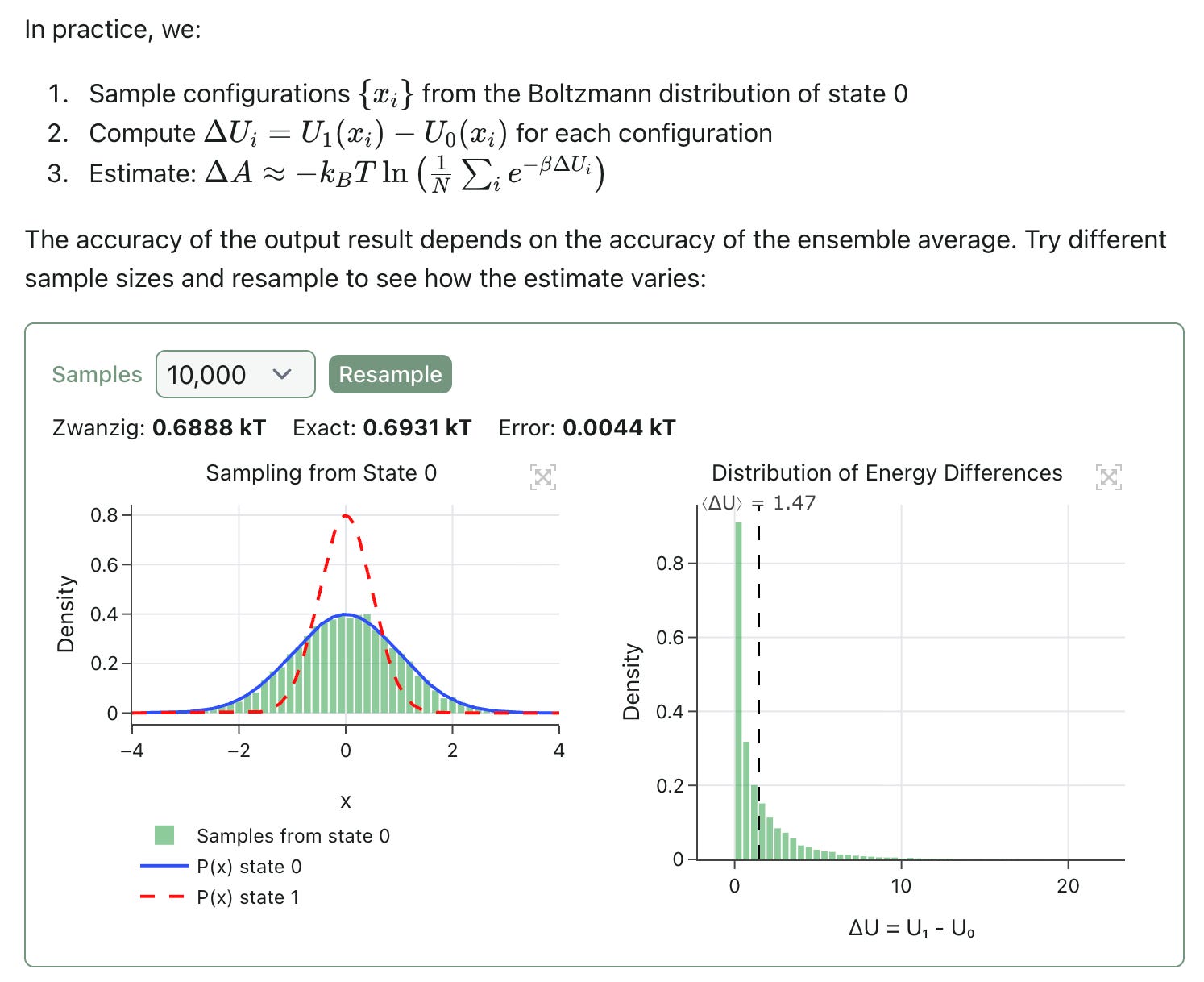
Free-energy perturbation (FEP) is a statistical mechanics–based technique for predicting the change in free energy between two states. The theory behind FEP is very interesting and much too complicated for this newsletter; as part of this launch, we’ve released an interactive blog post that makes it possible to explore some of the core concepts and build an intuition for what’s going on in an FEP calculation:
Most drug-discovery teams use FEP to predict the difference in binding affinity between related ligands: this is termed relative binding-affinity free-energy perturbation, or sometimes just RBFE for short. This matters because optimizing protein–ligand binding affinity is one of the crucial challenges of early-stage drug discovery. As a recent perspective by Mark Murcko highlights, the ability to identify compounds that bind a target strongly not only helps with dosage & selectivity but also gives drug designers more flexibility in optimizing biophysical properties & ADME. While there’s more to drug discovery than just binding affinity, drug designers broadly agree that knowing how modifications will impact affinity is quite useful.
Unfortunately, accurate computational prediction of binding affinity has proven very difficult. Simple single-structure methods like docking struggle to predict affinity with any predictive accuracy, while to date ML-based methods have struggled with generalization to new targets and chemical matter. FEP is noteworthy because it’s actually able to accomplish this task with high predictive validity, meaning that FEP can be used to effectively triage potential candidates in silico before needing to synthesize anything. FEP also doesn’t require existing experimental data for training, unlike QSAR-based methods, meaning that it can be used for new chemotypes or in other data-scarce regimes.
The ultimate dream of computer-assisted drug design is that scientists can design drugs in the computer, iterating quickly and cheaply to optimize their molecules without ever setting foot in the lab. While real life doesn’t quite work this way, FEP is about as close as our field has gotten to this goal—prospective studies show that compounds predicted to bind better by FEP often do bind better, with a success rate that’s substantially higher than other computational methods. When used at scale, FEP can lead to “startling efficiency gains in the overall hit-to-lead and lead optimisation process” (Chemistry World).
If all this is true, why isn’t FEP used constantly in preclinical discovery? Why doesn’t every medicinal chemist have a personal GPU running new FEP calculations around the clock to help them find the best binders possible? Two main reasons limit the broader diffusion of FEP into industry:
Expertise. Running FEP calculations properly is difficult and often requires specialized know-how. To get good results in a reasonable time, users must align the compounds properly, generate the graph of alchemical transformations to run, choose appropriate settings that balance speed and accuracy, actually run the calculations, and interpret the results—all of these are non-trivial for many potential users.
Computational cost. FEP is slow: one commonly cited figure is 10 GPU-hours per compound, although this obviously depends on a large number of factors. This runtime implies substantial real-world costs; a single FEP calculation can easily cost $20–30 worth of cloud compute time, if not more (in addition to software license fees).
While users and potential customers have been asking us about FEP for years, we’ve put this off until we thought we could offer meaningful improvements along these two dimensions. Today, we’re excited to launch a suite of tools and workflows to prepare, submit, and analyze FEP calculations that we think makes FEP dramatically faster & more accessible, with the ultimate hope of accelerating compound-discovery timelines for our customers.
Rowan’s Approach to FEP
Rowan’s FEP workflow is powered by TMD, a high-performance FEP engine built and maintained by Forrest York. TMD is based in part on the Time Machine engine developed at Relay Therapeutics, but has been extensively rewritten and optimized to achieve maximum performance on modern GPU hardware. TMD also incorporates many algorithmic improvements to improve performance and accuracy, including local resampling, adaptive lambda scheduling and Monte Carlo water sampling.
The quality of any FEP implementation is determined by accuracy (how well predicted binding affinities match experimental values) and speed (how long it takes to run the calculation). We’ve spent a good amount of time running benchmarks on Rowan FEP to try and understand how it compares to state-of-the-art methods. In our hands, Rowan FEP generally gives comparable accuracy to established methods, as assessed by both energy-based metrics (MAE and RMSE) and ranking metrics (Pearson and Spearman correlation).
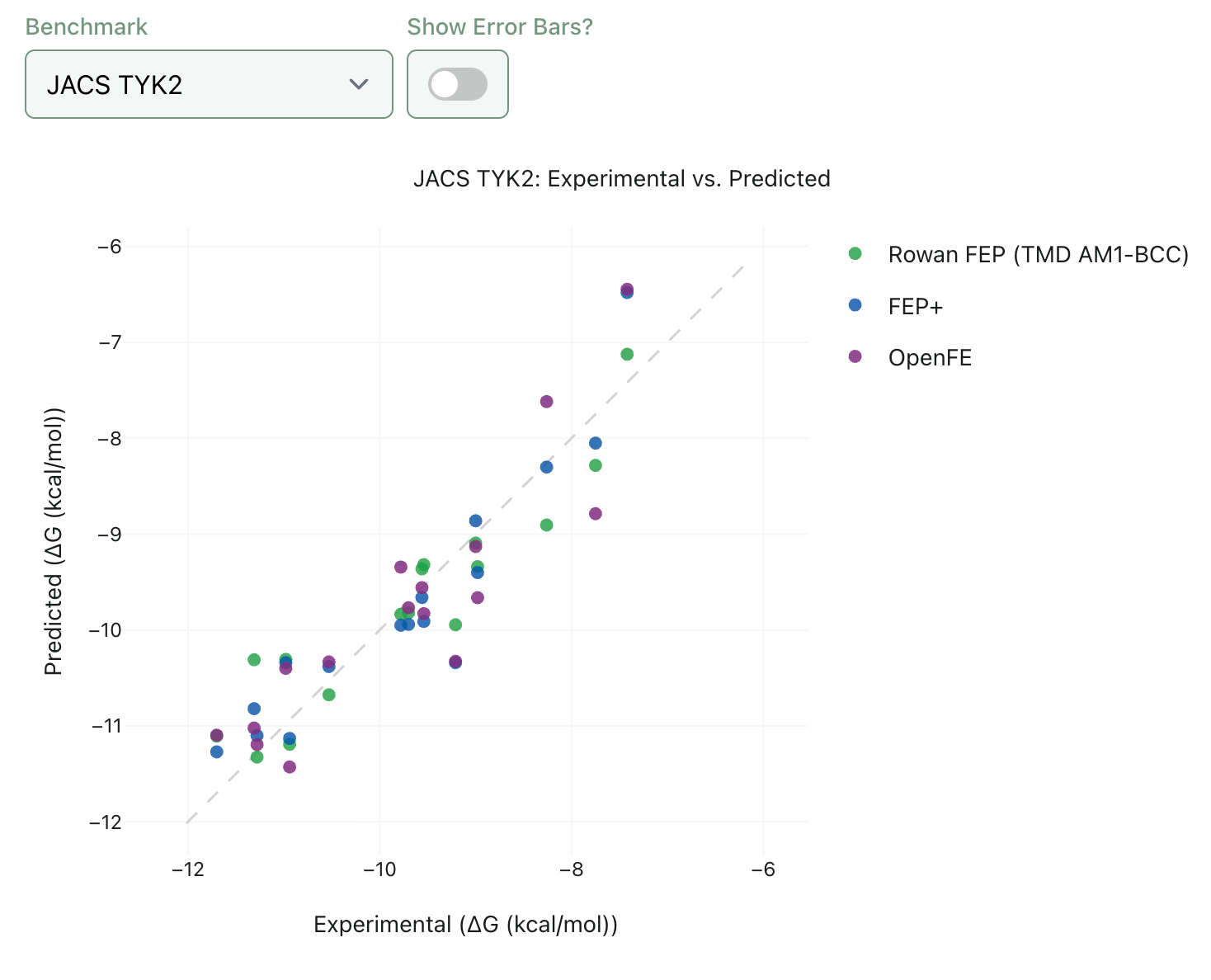
We want to be as transparent as possible about the accuracy and potential limitations of Rowan FEP, so we’re launching an entirely new section of our benchmark site with interactive plots of our FEP benchmarking data. If you’re curious, feel free to go check it out! We’ve added comparisons to FEP+ (Schrödinger’s proprietary industry-standard RBFE engine) and OpenFE (the leading open-source alternative).
Where Rowan FEP really excels, though, is speed. The myriad algorithmic improvements in TMD all make it dramatically faster to run calculations through Rowan FEP. While exact timings vary based on system and settings, Rowan FEP often takes only 10–20 minutes per alchemical leg, which translates to substantially higher throughput and lower costs for the end customer. (If you find this hard to believe, so did we! Timing results for TMD are also available on our benchmark site.)
Running FEP Through Rowan
When used properly, FEP can dramatically accelerate the compound-screening pipeline and improve timelines in lead optimization campaigns. But using FEP properly requires more than just a robust and performant engine like TMD: poses must be prepared and aligned properly, graphs have to be constructed, compute hardware has to be allocated, and so on.
We’ve divided the end-to-end FEP workflow into three high-level stages:
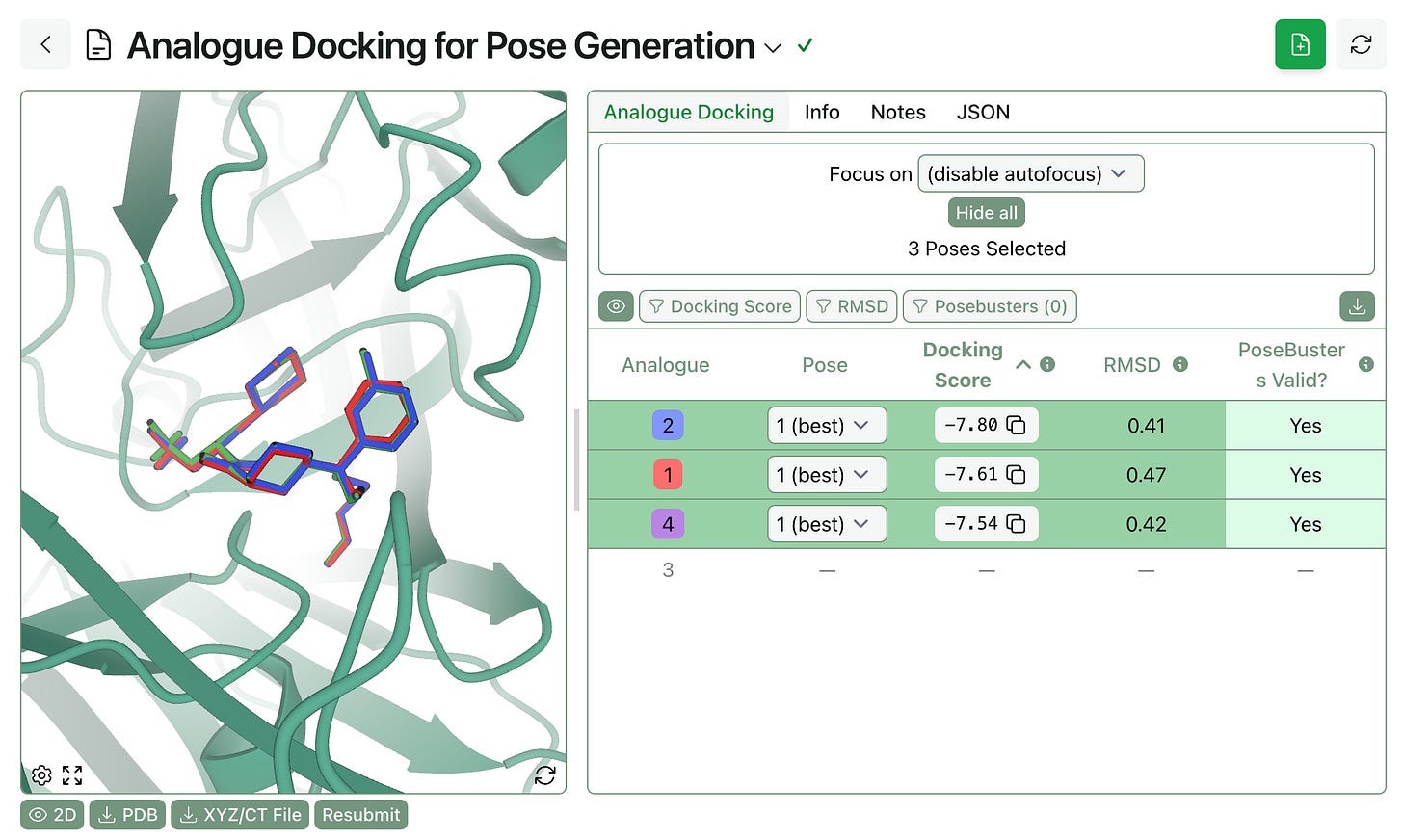
Pose preparation. FEP requires aligned input poses of the molecules under study. Rowan’s analogue docking workflow takes a protein, a template pose, and a list of analogues and uses MCS overlay to generate aligned protein–ligand structures suitable for FEP. Users can optionally filter by docking score, PoseBusters validity, or RMSD to the template.
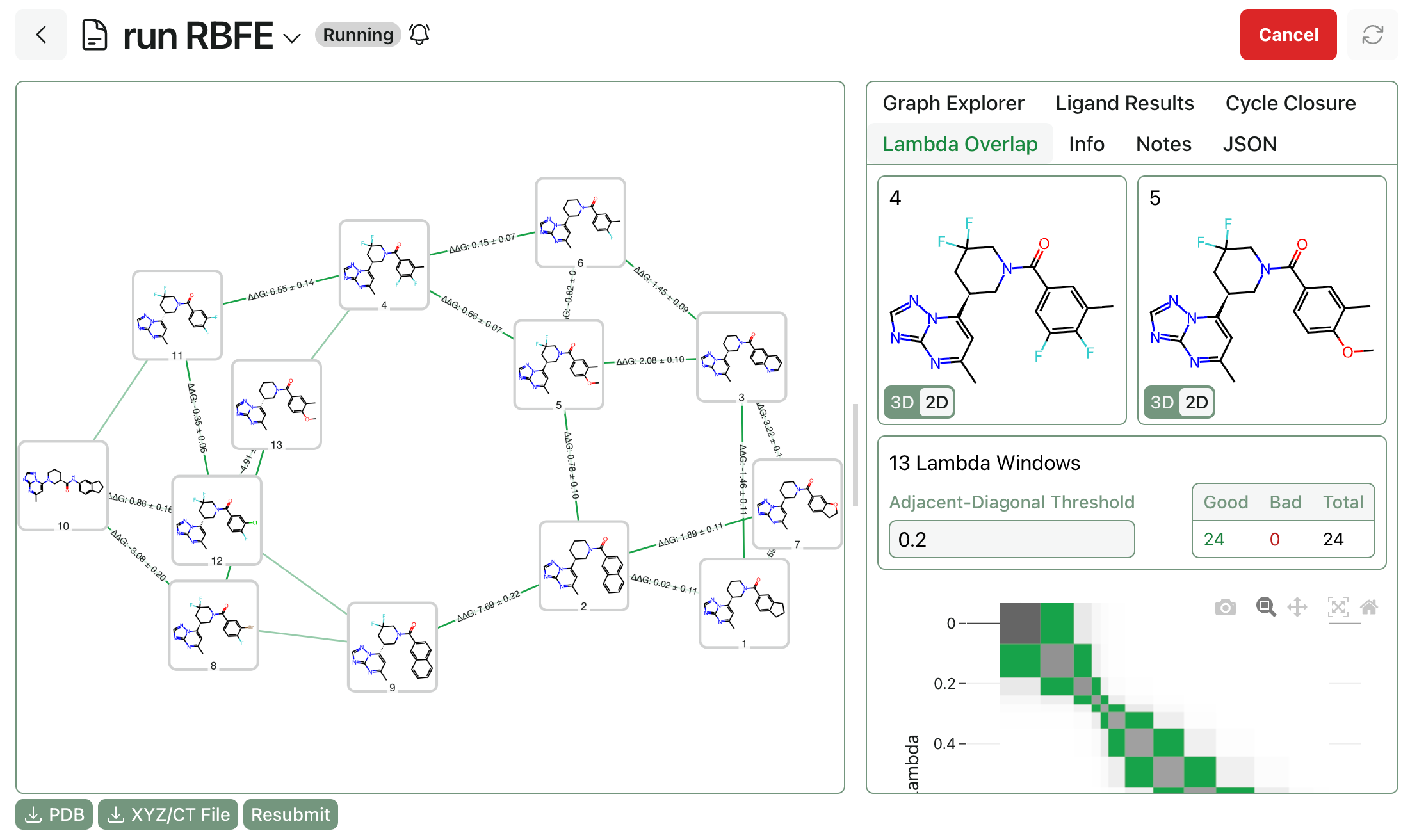
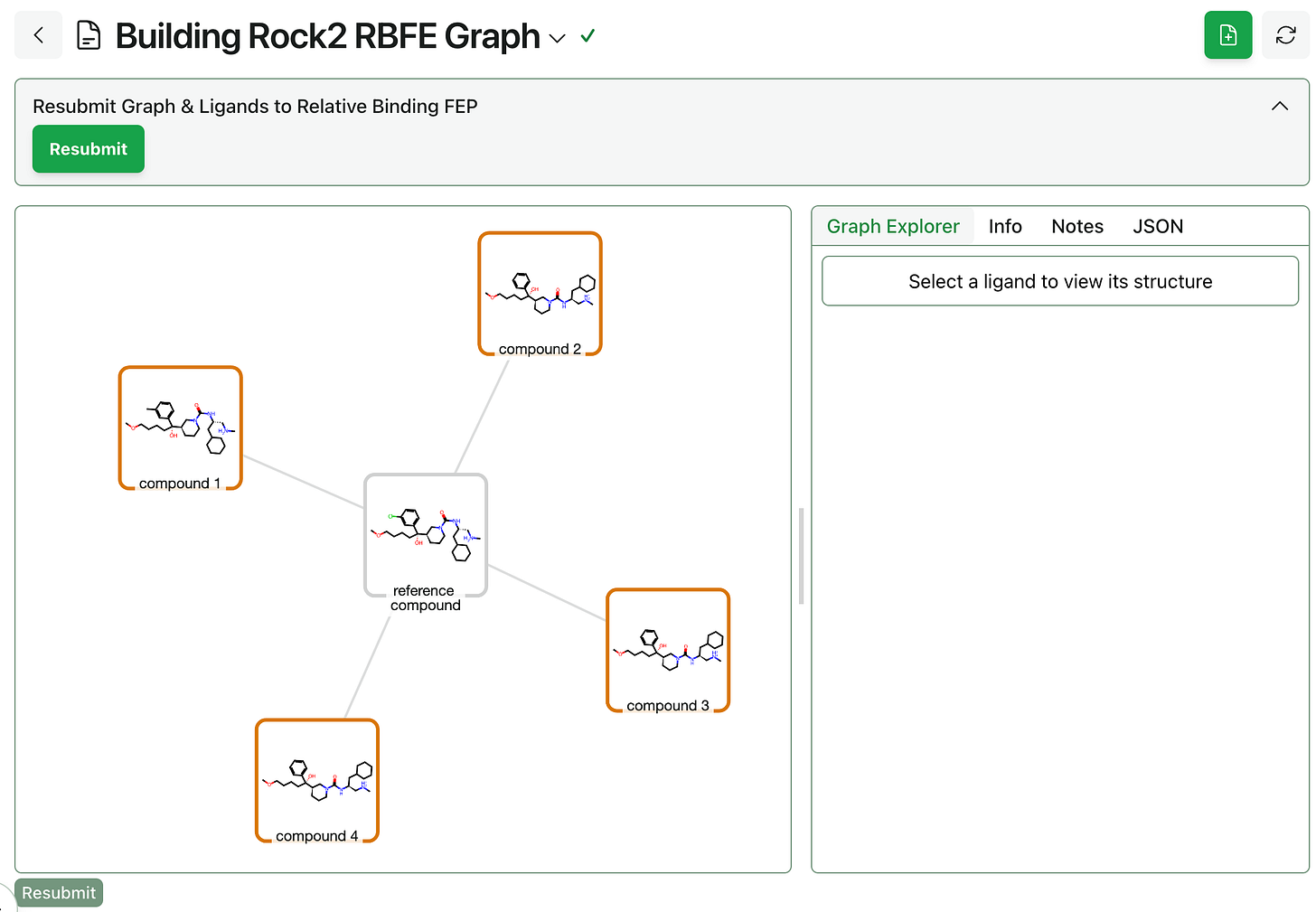
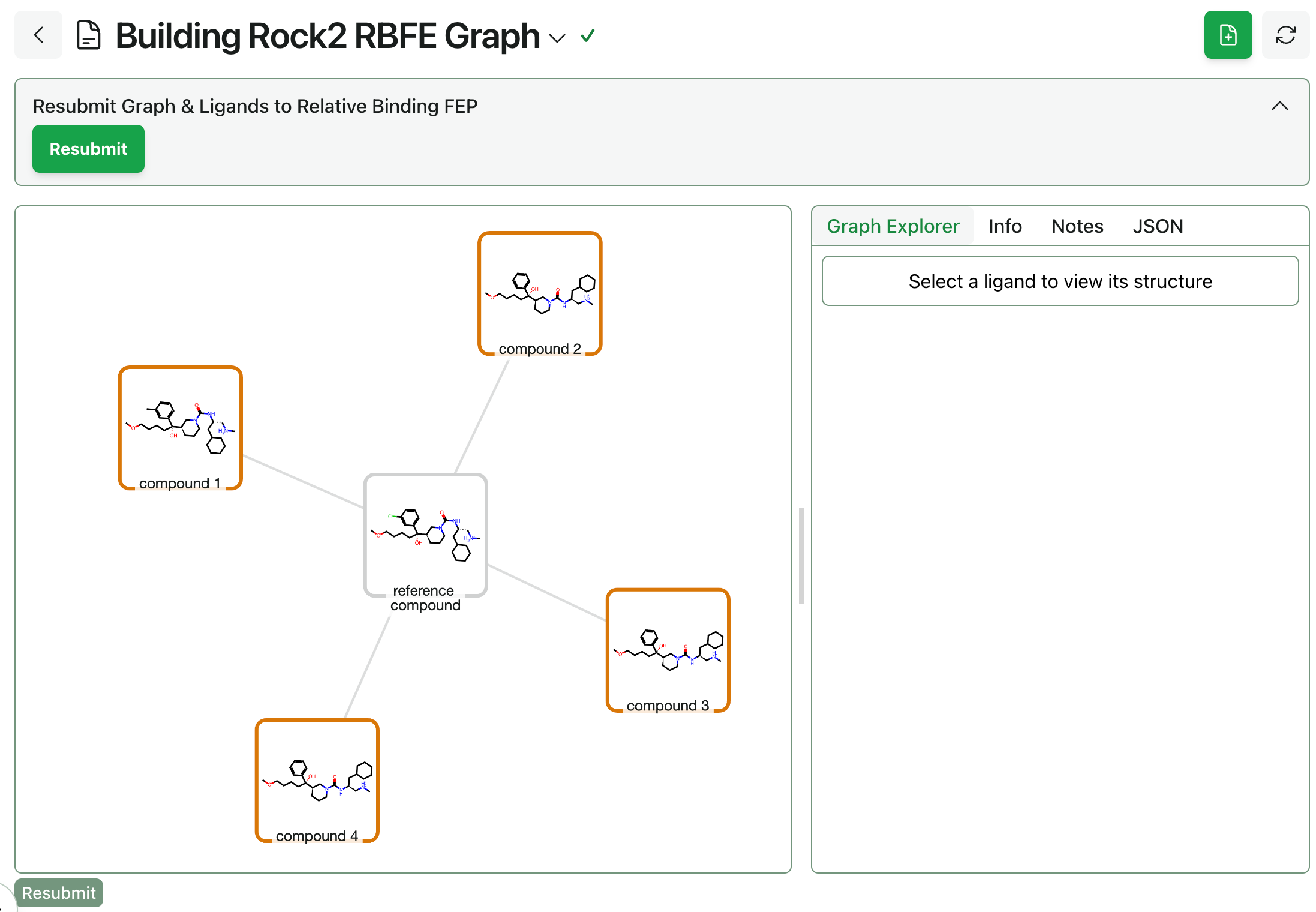
Graph construction. Next, the alchemical perturbation graph must be built through Rowan’s RBFE graph workflow. Users can choose between standard “greedy” mode and high-speed single-edge “star map” graph construction, as well as manually editing the graph once it’s been generated to adjust which edges will be computed.
Free-energy calculations. Finally, the RBFE calculations must be submitted and run. Users have full control over TMD settings, including simulation length, lambda overlap, and charge-assignment method, and sane default settings are provided for various use cases. Once submitted, Rowan will allocate cloud GPU resources and run the workflows, streaming per-edge results to the front end as edges complete.
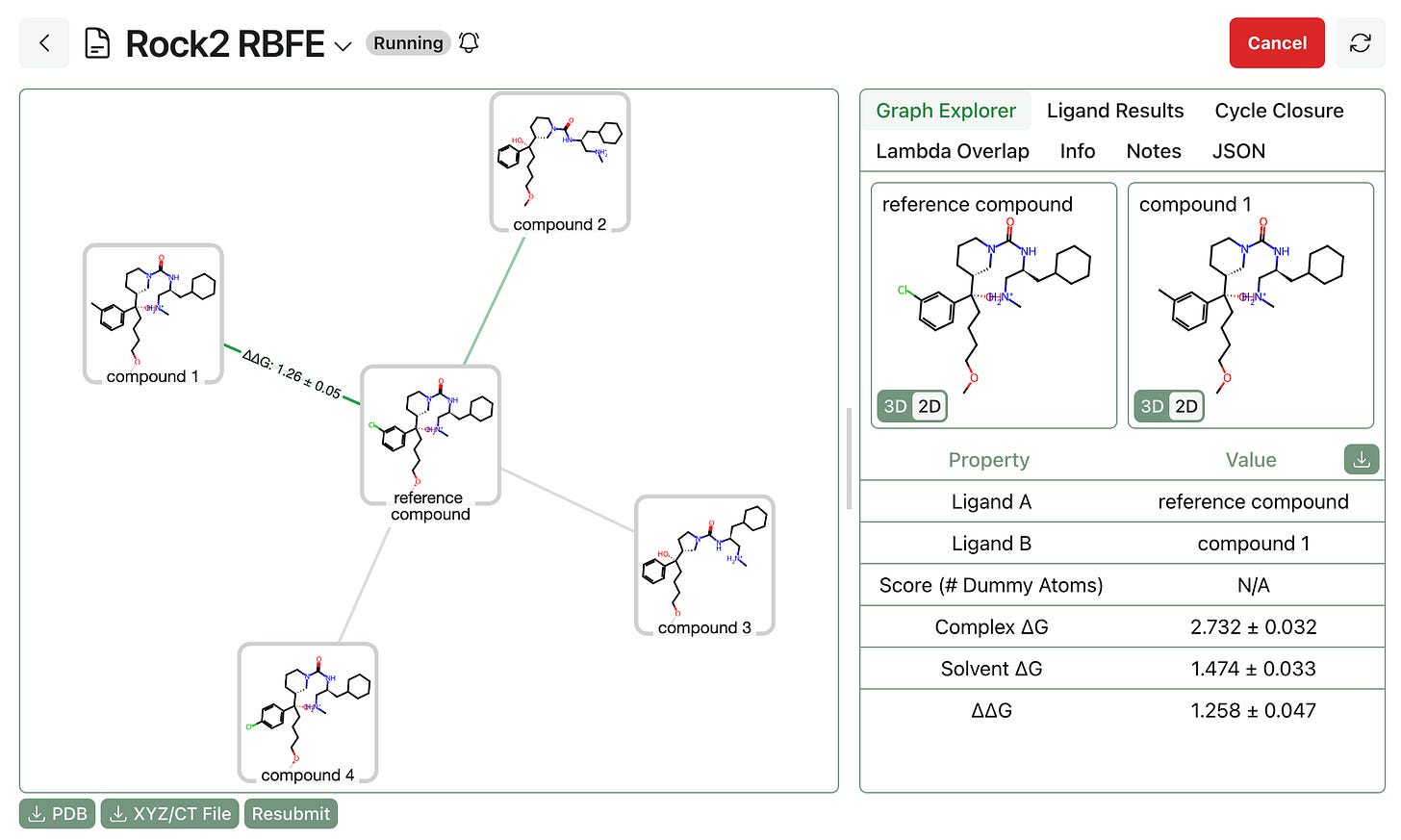
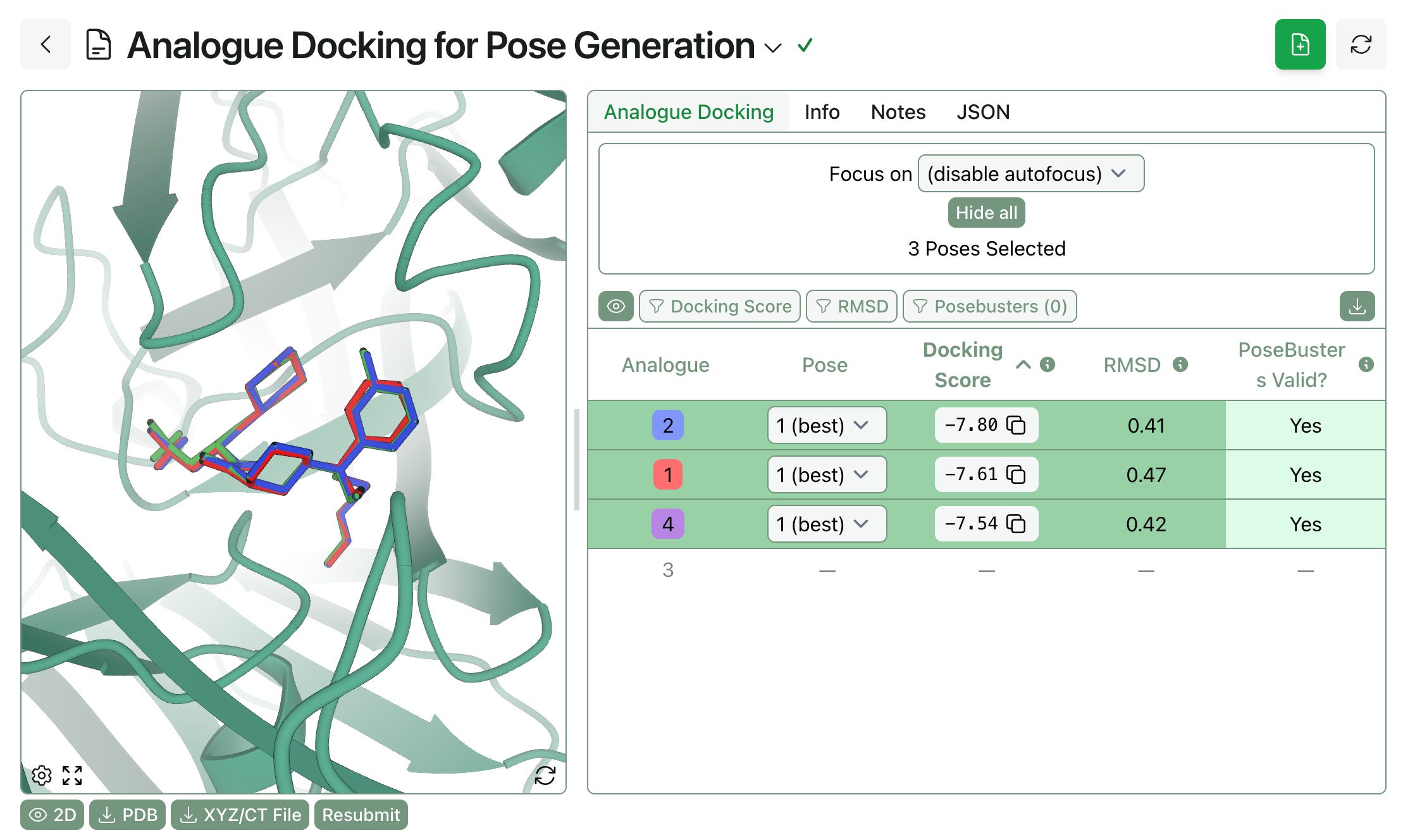
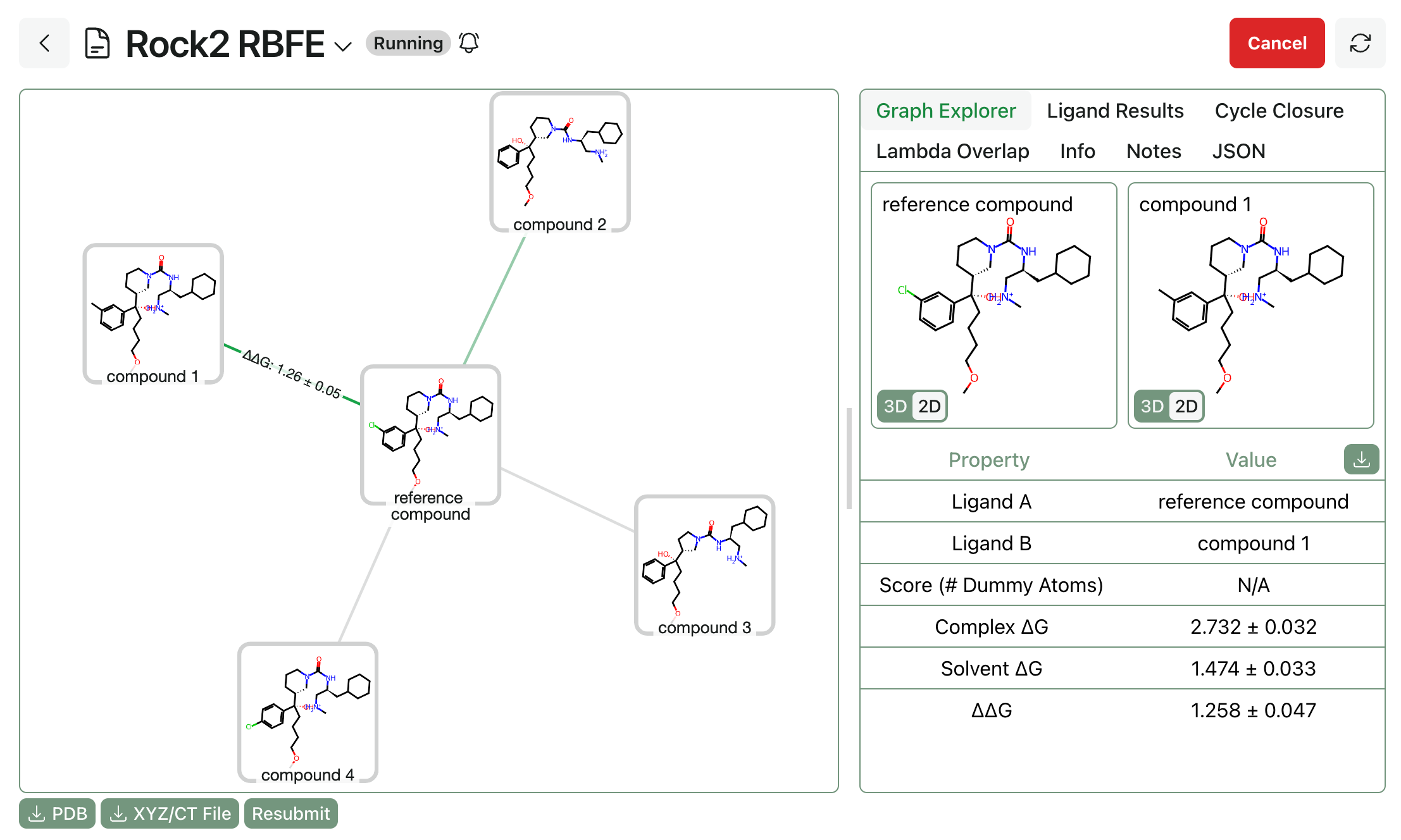
Once the whole workflow is complete, users can view final ligand rankings, per-edge lambda overlap values, cycle closure analysis, and trajectory snapshots.
We recognize that we’ve barely scratched the surface of the different ways that users can interact with FEP, tune the settings, and analyze their results. As always, we aspire to full scientific transparency: a full tutorial is available on our documentation site, as is an end-to-end video walkthrough.
The Potential of FEP
The high-level promise of FEP has always been to accelerate drug discovery by moving compound optimization into the computer (at least partly). While the core scientific challenges have been solved for about a decade, we think that the impact of FEP has been limited by the cost, complexity, and expertise needed to apply FEP to real-life drug discovery programs.
Rowan FEP aims to change this. While we’re not yet at a stage where FEP becomes “too cheap to meter” (borrowing a phrase from Sam Altman), we expect that lowering the cost of FEP will dramatically increase the impact we can have on early-stage drug-design programs. Making it affordable for drug hunters to run FEP on hundreds or thousands of compounds before every round of synthesis will increase the number of shots on goal and help design cycles converge more quickly—leading to better drugs, discovered faster.
We’re also hoping to make it possible for more chemists to submit, manage, and analyze these calculations for themselves. Rowan FEP provides scientists an easy-to-use interface for FEP while still giving users full control of the underlying science, removing the need for scientists to write code to run FEP workflows. In 2007, John Van Drie wrote that “today’s sophisticated CADD tools only in the hands of experts will be on the desktops of medicinal chemists tomorrow”; our goal is to fulfill this prediction for FEP.
Ultimately, we think low-cost FEP can change how early-stage drug discovery operates. One can imagine a world in which RBFE calculations are run around the clock for early-stage small-molecule programs, continuously exploring the SAR landscape around all new experimental hits and triaging ideas from medicinal chemists or generative algorithms. These in silico data can be used both to triage potential synthesis candidates and to provide input data for QSAR models that can be further refined by experimental data, helping to make sure that every compound synthesized is actually a “shot on goal.”
While this vision remains science fiction at present, we hope that we can get there. We’re excited to work with interested early adopters to explore integration of Rowan FEP into automated compound prioritization systems.
How to Use Rowan FEP
We’re releasing FEP in two ways today.
Organization Access
Organization-level customers (minimum $50K/year) of Rowan will have full access to FEP. There are no special usage caps or additional fees for FEP access; we will simply charge for FEP on a per-credit basis as we do for other workflows. (We expect that typical FEP usage with default settings will cost between $5–10 of credits per edge to start, and we hope to bring this down over time.)
As usual, single-tenant or VPC deployments are available for an additional fee; reach out to our team if this is of interest!
Rowan Managed FEP
We will also be trying out fee-for-service FEP modeling. In this paradigm, Rowan scientists will run FEP and directly return predictions to your team for a flat fee of $25/ligand. You can read more about how this works operationally on our Managed FEP page.
If you’re interested in working with us this way, reach out to schedule an introductory call with our team! We’ll discuss what structural data is available and what modifications you hope to study; if we both agree that the series is a good match for FEP, we’ll proceed.
(We’ve never done fee-for-service work as a business before. It’s possible that the way in which we structure these deals will change, or that we’ll stop doing this eventually—if you’re interested in working with us and have thoughts, please let us know!)
Although we’re happy to finally be releasing FEP after months of work and testing in private beta, we know our work is far from over. We’re excited to start deploying Rowan FEP and the rest of our drug-discovery tools into real early-stage discovery programs and working alongside scientific teams to optimize performance, improve reliability, and help discover drugs faster. If you’d like to partner with Rowan, reach out! We’ll be happy to talk.