
Fukui Indices & Global Electrophilicity
predicting regioselectivity + reactivity; applications to synthesis & metabolism; updates to our team



A common question in organic chemistry is that of regioselectivity: “where will my molecule react?” This is an important problem both in synthetic chemistry (predicting the site-selectivity of a given transformation) and in medicinal chemistry (predicting where a drug will be metabolized, e.g. by a P450 enzyme). Here’s an illustration of some of the most common transformations in Phase I drug metabolism—as you can see, there are a lot of potential sites of reactivity!

Unfortunately, regioselectivity is also tricky to predict. While in theory we could model each different pathway using quantum chemistry and compute ∆∆G directly, finding lots of transition states is time-consuming and not easily automated. For small molecules, it’s often possible to make good predictions simply through chemical intuition, e.g. by remembering that electron-donating groups direct electrophiles to the ortho or para positions in rings, but this becomes more challenging on large systems.
One of the more interesting solutions to this problem is “Fukui indices,” named after Kenichi Fukui (1981 Nobel Laureate in Chemistry), which quantify how a molecule’s electron distribution changes upon adding or removing an electron. To compute atom-centered (“condensed”) Fukui indices, you simply take the difference between the atomic charges of the neutral molecule and the corresponding anion or cation.
Experimentally, large Fukui indices are often found to correlate with sites of reactivity. There are three different Fukui indices for each atom (depending on whether the anion, cation, or both are included in the calculation), and each tells you something different: f(+) tells you where a molecule will react with nucleophiles, f(-) tells you where a molecule will react with electrophiles, and f(0) tells you where a molecule will react with radicals.
Parsing and visualizing Fukui indices can be a little time-consuming—to help, we’ve just released a workflow allowing for calculation of Fukui indices on Rowan. Here’s what f(-) looks like for 3-aminophenol—as you can see, the most nucleophilic sites are predicted to be (1) the amine itself, (2) the carbon para to the aniline, and (3) the carbon ortho to the aniline. Although the exact product ratio will of course depend on the properties of the nucleophile (hard–soft acid/base theory, etc.), this is still a useful first pass at predicting reactivity!

The ability to quickly predict Fukui indices is even more useful for larger molecules, where intuition is more difficult. In particular, Michael Beck has shown that in many cases f(-) can be used to predict sites of enzymatic metabolism in vivo. Here’s f(-) for phenprocoumon—experimentally, P450 hydroxylation is observed at the four sites highlighted in green, so the predictions are decent but not perfect. (There’s a substantial steric component to P450 hydroxylation that Fukui indices neglect, so this isn’t surprising.)

As a part of the same workflow, we’re also computing the global electrophilicity index. Global electrophilicity, computed as electronegativity squared divided by hardness, is “a quantity related to the capacity of an electrophile to promote a soft (covalent) reaction” (Maynard et al). A variety of papers support the ability of global electrophilicity to predict relative reactivity with soft nucleophiles (like reactive cysteine residues), making this parameter useful for the design of electrophilic warheads. A 2019 paper from Boehringer Ingelheim supports the ability of global electrophilicity to aid in the design of covalent warheads:
For acrylamides…the electrophilicity index of the educt correlates very well with the electrophilicity index in the transition state and the electrophilicity index in the transition state geometry then correlates with the activation energy. This allows to predict the reactivity of a molecule solely from the electrophilicity index in the educt geometry. As no transition state has to be calculated this greatly reduces the computational time requirements to estimate the activation energy.
When applied with appropriate measures, the warhead associated electrophilicity index is a valuable tool for a computer aided drug design scientist, which gives insight into the reactivity of covalent inhibitors.
We can gain some intuition for global electrophilicity with a practical example. Here’s a Rowan calculation on N-(p-cyanophenyl) acrylamide. Visualization of f(+) shows potential sites of nucleophilic attack—as expected, the beta carbon and the carbonyl carbon are highlighted.
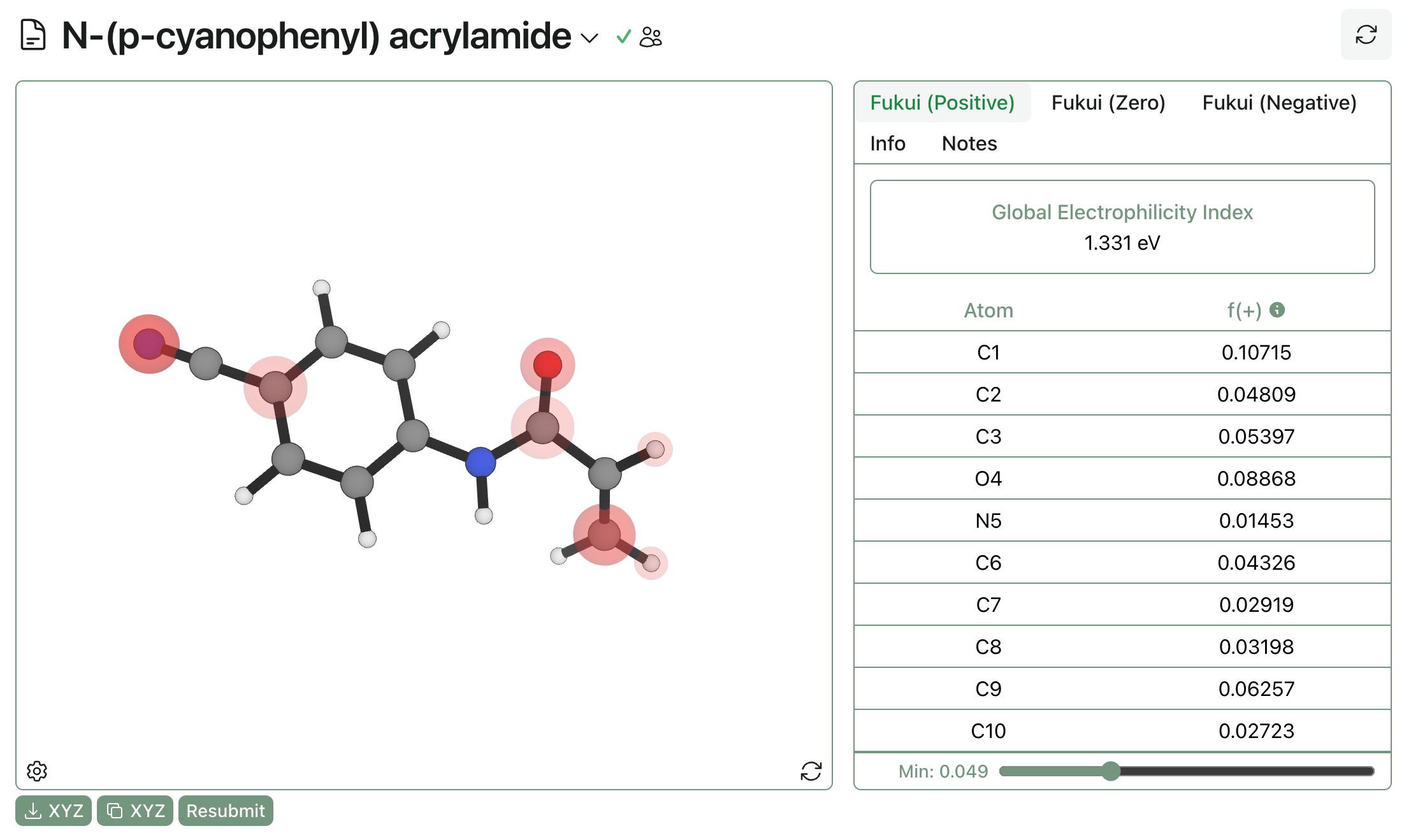
The global electrophilicity index is 1.331 eV, which becomes more meaningful when compared to other acrylamides: N-phenyl acrylamide has a GEI of 0.944 eV, and N-(p-methoxyphenyl) acrylamide has a GEI of 0.844 eV. As expected, making the ring more electron-poor increases the electrophilicity and the reactivity of the acrylamide. While the trend is pretty intuitive in this case, the ability to generate quick predictions can be quite useful in more complex cases (like scaffold hops or similar).
Our new workflow runs extremely quickly—the above calculation took only nine seconds—so we anticipate that this can be a valuable conceptual tool for anyone thinking about designing or modulating reactivity and selectivity in small molecules. Try it out today!
Finally, we’re thrilled to welcome Jonathon Vandezande to Rowan as our Director of Computational Chemistry! Jonathon joins us with experience from Schrödinger and the Max Planck Institute for Kohlenforschung, previously completing a PhD in quantum chemistry at the University of Georgia.