


Hydrogen-Bond-Basicity Prediction Made Easy
not all hydrogen-bond donors are created equal; the pKBHX scale; predicting pKBHX in Rowan; case studies & a preprint

 – Art & Theology")
Hydrogen bonding is one of the most fundamental interactions in chemistry: hydrogen bonds hold liquid water together (see above image, which has water in it) and are important structural elements in virtually every protein–ligand complex. But while most practicing organic chemists have plenty of pKa values or bond-dissociation energies memorized, the strength of various hydrogen-bond donors or acceptors is much less appreciated.
The strength of different hydrogen-bond acceptors can be quantified through pKBHX, the base-10 logarithm of the equilibrium constant between the acceptor and a given hydrogen-bond donor. For historical and pragmatic reasons, it’s common to use 4-fluorophenol as the hydrogen-bond donor and carbon tetrachloride as the solvent; these choices are somewhat arbitrary, but values measured under these conditions correlate pretty well with pKBHX values measured with other hydrogen-bond donors or in other solvents. Under these conditions, pKBHX values for common functional groups span about six orders of magnitude, from –1 to 6:
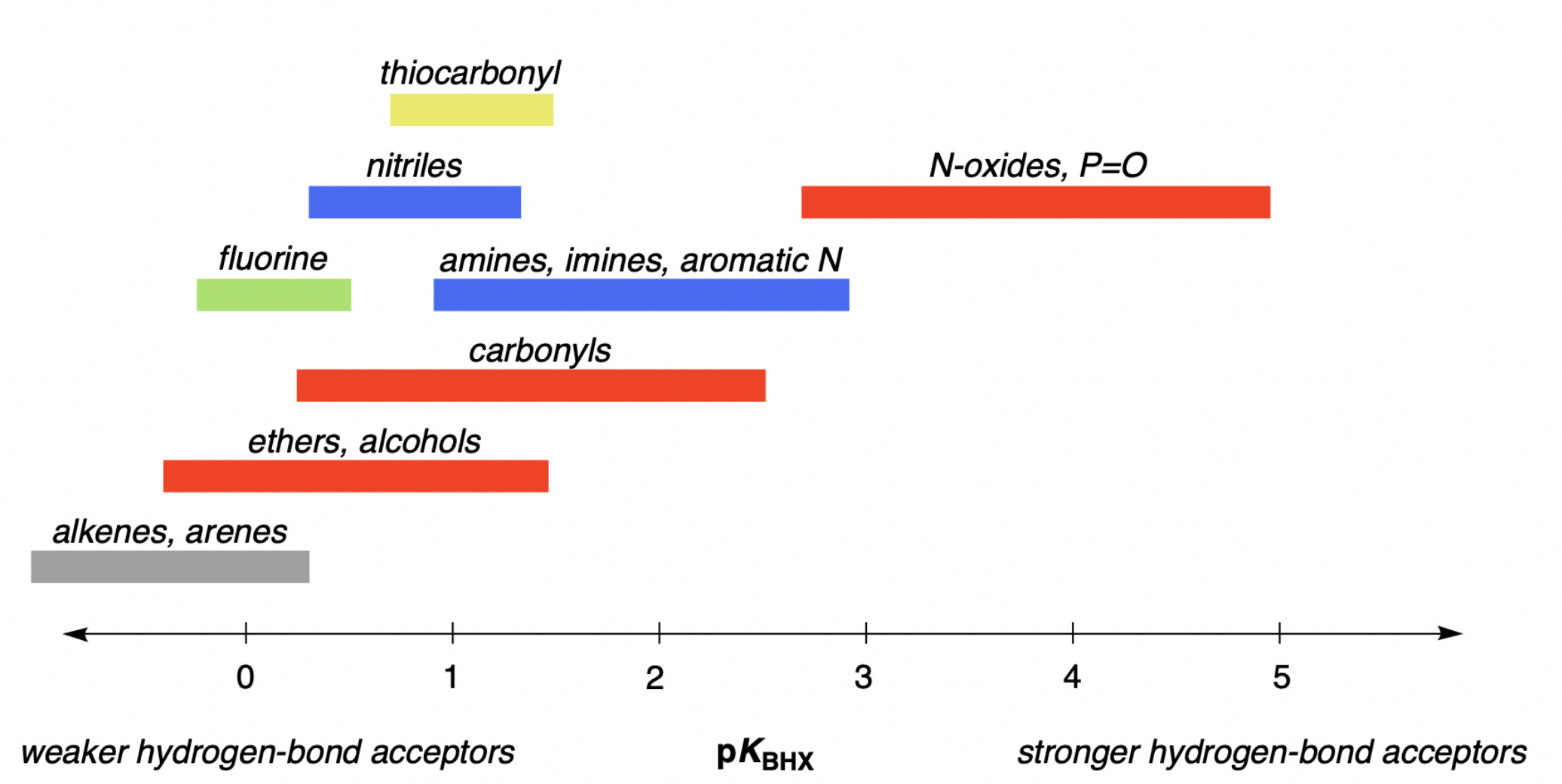
While hundreds of pKBHX values have been measured under these conditions, there’s still a need for computational methods capable of accurately predicting pKBHX values. Tons of different pKBHX-prediction workflows have been developed over the past three decades, but despite all this research there’s still not a robust solution for arbitrary small molecules that a regular chemist can use for their research.
Today, we’re excited to address this issue by releasing Rowan’s pKBHX workflow (preprint, ChemRxiv). To predict pKBHX values, we locate electrostatic potential minima in the region of hydrogen-bond acceptors, which indicate regions of electron density, like lone pairs. Here’s a visualization of the –0.04 EH/e electrostatic potential isosurface for a model drug-like molecule—you can see that the hydrogen-bond acceptors have blobs of negative electrostatic potential near them.

We locate the electrostatic potential minima near each atom via numerical optimization, and linearly scale the values to match experimental pKBHX values. This approach was first proposed by Peter Kenny and co-workers—we’ve sped it up by using the AIMNet2 neural network potential for geometry optimization and running only a single low-cost r2SCAN-3c calculation per molecule, avoiding the conventional time-consuming geometry optimization with DFT. Our workflow gives a mean error of 0.19 pKBHX units on a published experimental database, similar with previous workflows.
One advantage of Vmin-based pKBHX prediction is that we not only predict the numerical strength of different hydrogen-bond acceptors, but also the potential geometry of hydrogen bonds. For instance, here are the predicted hydrogen-bond-acceptor sites for the above compound—while hydrogen bonds to the pyrazole and the pyridazinone are predicted to be linear, the amide is predicted to have two separate hydrogen-bonding sites, one of which is significantly stronger than the other.

These predictions aren’t just intellectually interesting; they’re also practically useful. Modulating the strength of hydrogen-bond acceptors can have dramatic impacts on the lipophilicity, permeability, efflux ratio, and binding affinity of potential small-molecule drugs.
Here’s a case study from Sébastien Degorce and coworkers at AstraZeneca: while trying to optimize the permeability of IRAK4 inhibitors as treatments for diffuse large B-cell lymphoma, they modified the core pyrrolopyrimidine scaffold (A.14) and found that different modifications had dramatic impacts on the compound’s properties. Switching the position of the nitrogen in the pyrrole ring (A.17) made the compound more hydrophilic (logD7.4 ↓), decreased membrane permeability (Caco2 Papp ↓), and increased efflux (efflux ratio ↑)—all very bad changes if the goal is an orally bioavailable drug! In contrast, switching to a pyrrolotriazine scaffold (A.20) increased lipophilicity, increased permeability, and decreased efflux.

These changes might seem random, but they can all be understood in terms of hydrogen-bond-acceptor strength. Compound A.14 has two moderately strong hydrogen-bond acceptors (pKBHX 2.03 & 1.47), and moving to the other pyrrolopyrimidine scaffold (A.17) increases their strength dramatically (pKBHX 2.64 & 1.62). It’s worth noting that pKBHX is a log scale, just like pKa, so an increase of 0.61 pKBHX units corresponds to a 4x increase in hydrogen-bond basicity. In contrast, moving to pyrrolotriazine (A.20) lowers the pKBHX values to 1.31 and 1.36, a 5x decrease in hydrogen-bond-acceptor strength for the bottom nitrogen.
There’s lots more that we could say about this topic—but we’ll defer interested readers to our preprint, with many more case studies and a more detailed discussion. Subscribing Rowan users can access the hydrogen-bond-basicity workflow as of today—happy computing!