
ML Models for Aqueous Solubility, NNP-Predicted Redox Potentials, and More
the promise & peril of solubility prediction; our approach and models; pH-dependent solubility; testing NNPs for redox potentials; benchmarking opt. methods + NNPs; an FSM case study; intern farewell




Apologies for the surprise second newsletter this week, but we have more to release! Since Wednesday, we’ve released two new solubility-prediction methods, a way to predict pH-dependent solubility, two new benchmarks, and a video tutorial on how to use Rowan’s double-ended transition-state-search workflow.
Predicting Aqueous Solubility and pH-Dependent Solubility
Since humans are mostly water, almost all drugs must dissolve in water to reach their target. Predicting the aqueous solubility of potential drugs is an important task in drug discovery; unfortunately, it’s also quite difficult to do well. There are literally hundreds of papers proposing various physics-workflows, cheminformatic methods, and ML models for predicting aqueous solubility, but it’s hard to know which ones work well. Pat Walters has a fantastic blog post called “Predicting Aqueous Solubility - It’s Harder Than It Looks” in which he discusses the issues with most benchmarking work, including data leakage and poor statistics.
Here at Rowan, we’re quite interested in aqueous solubility prediction because we know how important getting this right will be for our customers. Inspired by Pat’s post, we used a recent high-quality dataset to benchmark a wide range of methods and training objectives, ranging from simple multiple linear regression-based approaches to complex pre-trained neural network potential-based approaches. We used Butina splitting to minimize data leakage and used a variety of external test sets to compare performance. (Kudos for Eli for training literally hundreds of models here!)
We found that a variety of approaches performed pretty well in our hands. Here’s the performance on our Butina-split test set, for instance:
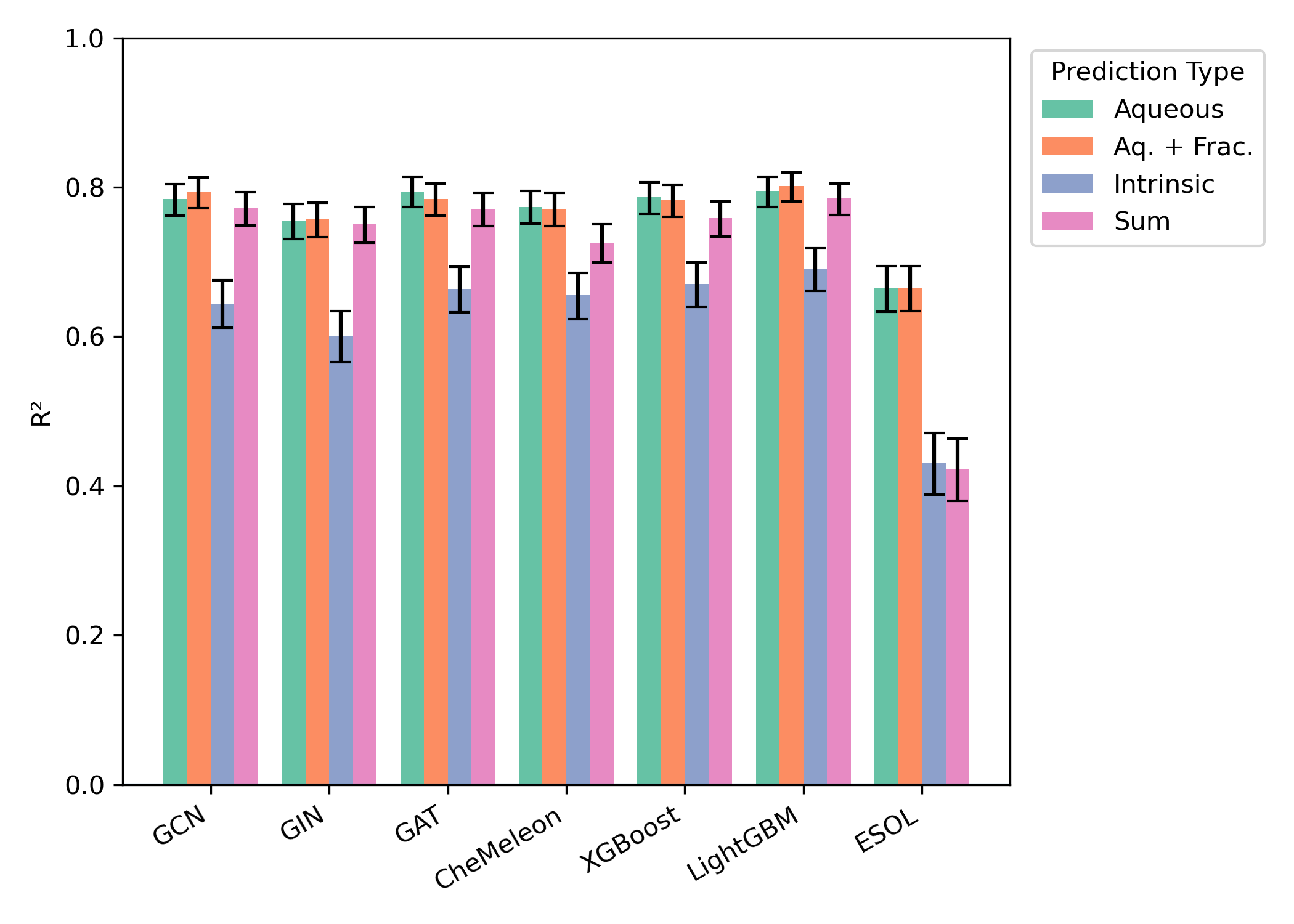
In combination with our macroscopic pKa workflow, we also found that we could predict pH-dependent aqueous solubility in good agreement with experimental values:

For more methodological details and way more data, read our paper here!
New Rowan Features
As a result of this work, we’re adding three new capabilities to Rowan:
(1) All Rowan users will be able to predict aqueous solubility using our implementation of the venerable ESOL model (which we re-fit on our dataset). Although ESOL is simple, it will be considerably more accurate than fastsolv for aqueous solubility.
(2) Rowan subscribers will also be able to use our ML “Kingfisher” model to predict aqueous solubility. (If you’re interested in seeing where Kingfisher is better than ESOL, check out our paper!)
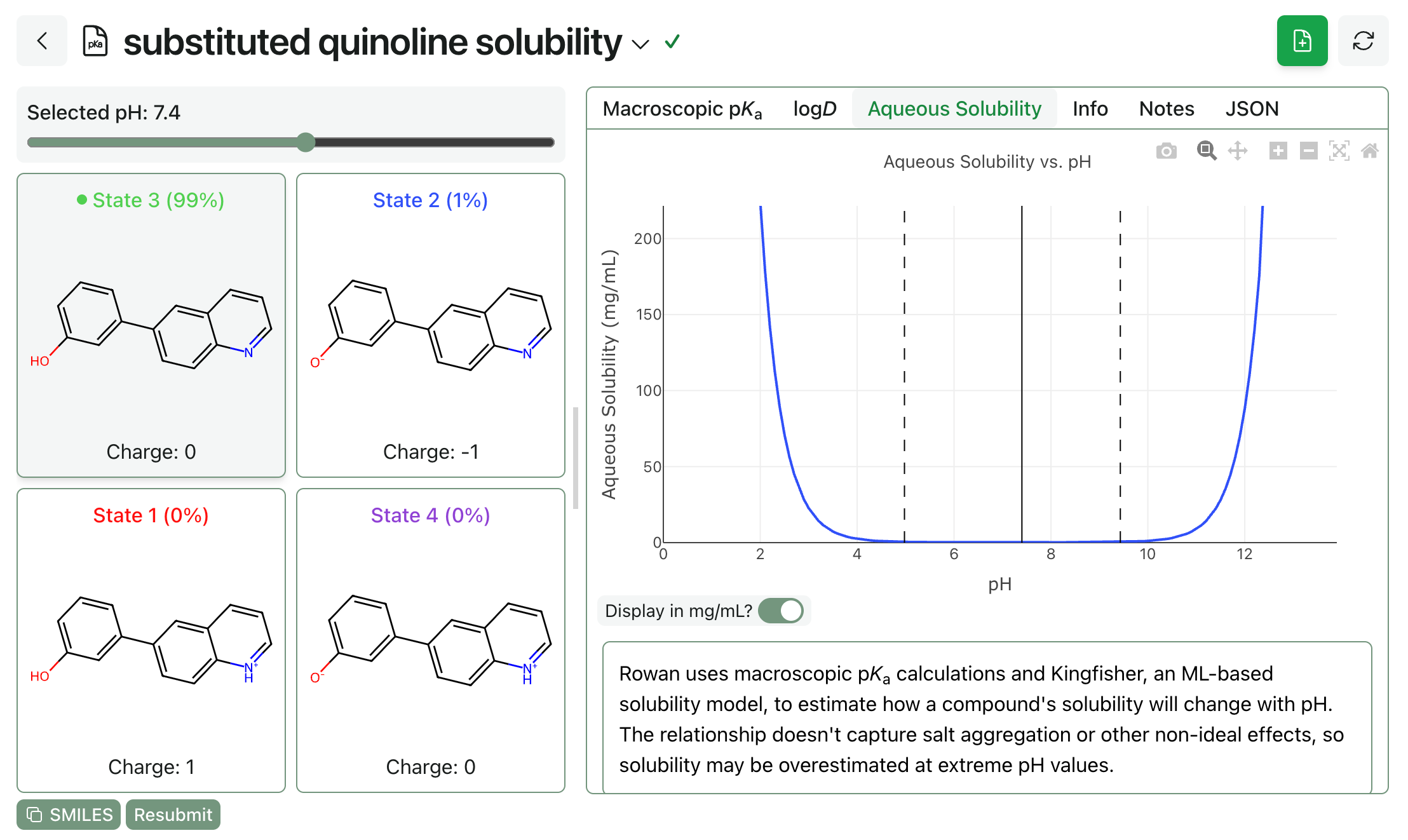
(3) And Rowan subscribers will also be able to predict pH-dependent aqueous solubility through the macroscopic pKa workflow. Here’s what this looks like:
Benchmarking OMol25-Trained NNPs For Redox Potentials and Electron Affinity
The latest generation of neural network potentials are able to predict charge- and spin-dependent energies across the periodic table, which in theory means they can be used to predict properties like redox potentials and electron affinities. Since these models don’t actually encode the explicit physics behind charge and spin, though, one might expect that they’d be pretty bad at these properties.
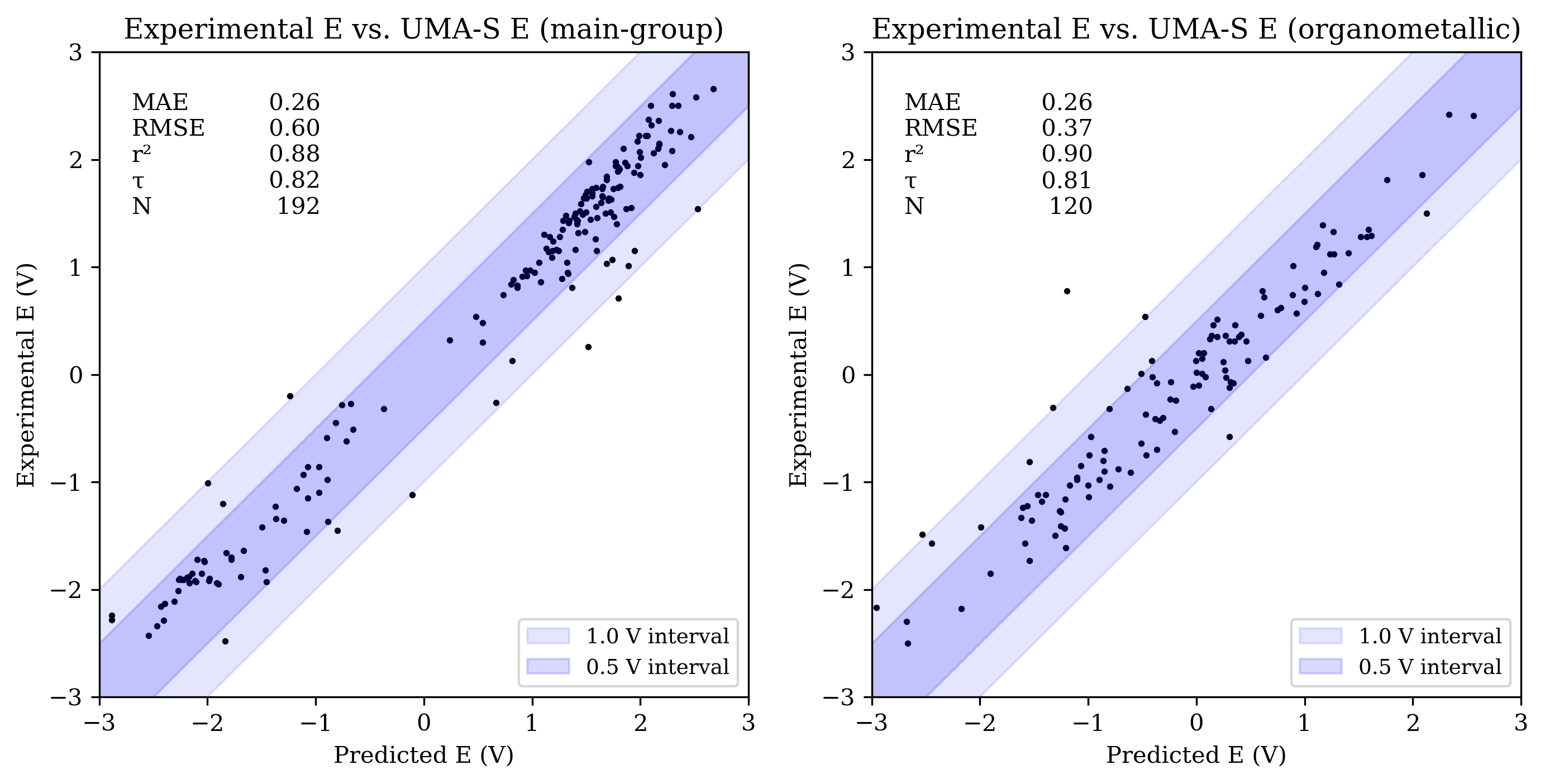
Our intern Sawyer VanZanten benchmarked UMA-S, UMA-M, and eSEN-OMol25-sm-conserving against a variety of datasets to answer this question. The results are very interesting! While these NNPs aren’t clearly better than conventional low-cost DFT methods for organic systems, they seem to avoid the pathologies of low-cost methods for organometallic species (perhaps because they inherit the accuracy of the high-level DFT training data). Qualitatively, the results are very reasonable and certainly good enough to be useful for exploratory screening of electronic properties. You can read Sawyer’s full writeup here.
Here’s the UMA-S-predicted redox potentials of Grimme’s ROP313 dataset vs. experimental values:
Which Optimizer Is Best For NNPs?
As a part of our benchmark site, benchmarks.rowansci.com, we’ve been testing the ability of different NNPs to optimize regular drug-like organic molecules. We used Sella by default, but following the OrbMol release there was a bit of discourse on X about which optimizer is best for this use case.
In the spirit of scientific exploration, we decided to conduct a little benchmark of different optimizers and NNPs. We found that Sella works quite well and that almost all methods struggle to converge to true minima without any imaginary frequencies. If this is interesting or relevant to you, check out the full blog post! We discuss the limitations of this study, practical implications, and directions for future research.
Using the Freezing-String Method to Study Azide–Alkyne Cycloaddition
In our last newsletter, we shared several case studies about how Rowan’s new double-ended transition-state-search workflow could be used to find interesting or non-trivial transition states. To illustrate what this process looks like end-to-end, we’ve recorded a video walkthrough of all the steps needed to run a transition-state search for an azide–alkyne cycloaddition, optimize the transition state, and confirm its identity with an intrinsic-reaction-coordinate calculation. Check it out on Youtube!
Another Farewell to Interns
Several weeks ago, we bid our interns Vedant and Ishaan farewell. They shared their parting reflections, notes on the cultural differences between industry and academia, and tips for GeoGuessr—read the previous writeup here.
This week, we say goodbye to Isaiah and Sawyer, our other two interns. Isaiah led Rowan’s work exploring how computational chemistry can be used to improve chemical education, while Sawyer did the aforementioned redox-potential-benchmarking work and a few other unreleased projects. Read their reflections and GeoGuessr tips here.

Until next time, happy computing!