
openconf and other Open-Source Projects
enabling & being enabled by open science; lacunæ in open-source conformer generators; a fast Monte Carlo–powered solution; macrocycles; obtaining topologies from 3D coordinates; fast Butina splitting





Rowan is a business that’s enabled by open-source scientific code. Packages like the RDKit, xTB, GPU4PySCF, and TMD (among others) are core to what we do, and we aim to give back to the open-source community both by contributing to existing open-source projects and creating new ones where we perceive gaps.
We’ve previously shared about some of the open-source projects that we’ve developed (Egret, PyPermm), help maintain (CoSIMS), or contribute to (ML-FSM). Today, we want to highlight a few of the new open-source projects we’ve been working on recently: a new conformer-generation package (”openconf”), a method for converting XYZ coordinates into RDKit molecules (”steamroll”), and a fast way for Butina-splitting large datasets (”Chalcedon”).
openconf: Rapid Monte Carlo–Based Conformer Generation
Most complex molecules can populate many different conformations, and finding the relevant conformers for new molecules is a big problem in molecule simulation. To date, Rowan has mainly relied on two methods for conformer generation:
ETKDG, implemented in the RDKit, uses an experimental distance geometry–based method to embed molecules into 3D space based on their topology. (There are a few related variants here: KDG without experimental torsional information, ETKDGv3 which performs better for small rings & macrocycles, and so on.)
iMTD-GC, implemented in CREST, uses iterated metadynamics and genetic crossing in combination with xTB methods to sample configurational space before identifying distinct minima.
While we’re big fans of both of these methods (and the science behind them), neither is perfect for all of our use cases at Rowan.
ETKDG often misses conformers or generates high-energy conformers for large systems (e.g. PROTACs) and has additional random pathologies (e.g. a curious preference for twist boats over chairs). While these problems can be ameliorated by generating thousands of conformers with ETKDG and then deduplicating, this can become quite slow (and a large majority of the replicas often converge on the same few conformers).
In contrast, iMTD-GC works well if you use quite careful settings, but struggles to generate high-quality ensembles for drug-like molecules with “quick” settings. Additionally, we’ve often run into issues with segfaults and other stochastic system errors in CREST, which creates issues when running calculations at scale through Rowan.
After a few years of fighting these issues and struggling to robustly get good performance in high-throughput contexts, we decided to bite the bullet and build our own conformer-generation method. Rather than trying to completely replace ETKDG or iMTD-GC for all applications, we aimed to quickly generate diverse and reasonable conformer ensembles for drug-like molecules that we could use in downstream applications like docking, pKa prediction, strain estimation, and so forth.
The result of this project is openconf, an open-source Monte Carlo–based conformer-generation package that builds atop the RDKit to provide physically reasonable conformer ensembles as quickly as possible. While we’re not the first people to explore Monte Carlo as a way to generate conformer ensembles (far from it), we think openconf is a nice addition to the modern open-source conformer-generation landscape. It’s written in modern Python, has only a few dependencies, and can be easily installed using your favorite Python package manager.
How openconf Works
openconf uses Monte Carlo moves to quickly explore conformational space. To ensure that reasonable conformers can be found even for complex systems, openconf implements a variety of Monte Carlo moves:
Single- and multi-torsion: rotate around one or more rotatable bonds
Correlated torsion: rotate a few adjacent bonds simultaneously
Global shake: move a majority of the rotors all at once to escape local minima
Ring flip: reflect e.g. a cyclohexane ring from one chair conformation to its mirror image
Crankshaft: rotate an arc of ring atoms around the axis connecting two anchor atoms
Kinematic inverse closure: rotate a macrocyclic ring bond and then uses cyclic coordinate descent to solve for the remaining ring torsional angles.
Macrocyclic amide flips: convert ring amides between cis and trans conformers and re-close the rest of the ring (this becomes important for cyclic peptides)
The main openconf loop takes a pool of ETKDG-generated seeds and adds new conformers to the pool by iteratively applying the above moves and optimizing the output conformers using MMFF94. The success rate of each move is tracked, allowing openconf to adaptively adjust move probability, and convergence is tracked to enable early stopping for simple systems. (Ongoing deduplication of the conformer pool is key here; to improve performance, we’ve added batching to PRISM Pruner that makes deduplication c. 15x faster.)
openconf also works for inorganic and organometallic complexes, including lanthanides: UFF is used instead of MMFF94 and restraints are added to prevent metal–ligand dissociation. (Performance is still a bit flaky for some inorganic complexes; we hope to improve this in the future.)
Performance
In practice, openconf generates large and diverse ensembles more quickly than either ETKDG or iMTD-GC. While openconf is slower than ETKDG at generating 10 conformers, since the pool and seed overhead dominates, openconf becomes noticeably faster at 50+ conformers. Benchmarking on experimental datasets like OpenEye Iridium also confirms that openconf matches or outperforms ETKDG at the same budget for flexible drug-like molecules.
We’ve already been using openconf within Rowan for generating FEP input poses (see our previous newsletter), and we’ve now integrated openconf into many more parts of our platform. For Rowan’s physics-based pKa methods (employing AIMNet2 and g-xTB), we’ve found that openconf leads to lower errors and noticeably faster performance than either ETKDG or iMTD-GC. On a sample set of 16 drug-like molecules, openconf-based pKa prediction was over 4x faster than ETKDG-based pKa prediction (190 s for openconf vs 846 s for ETKDG) while giving lower mean error and fewer outliers.
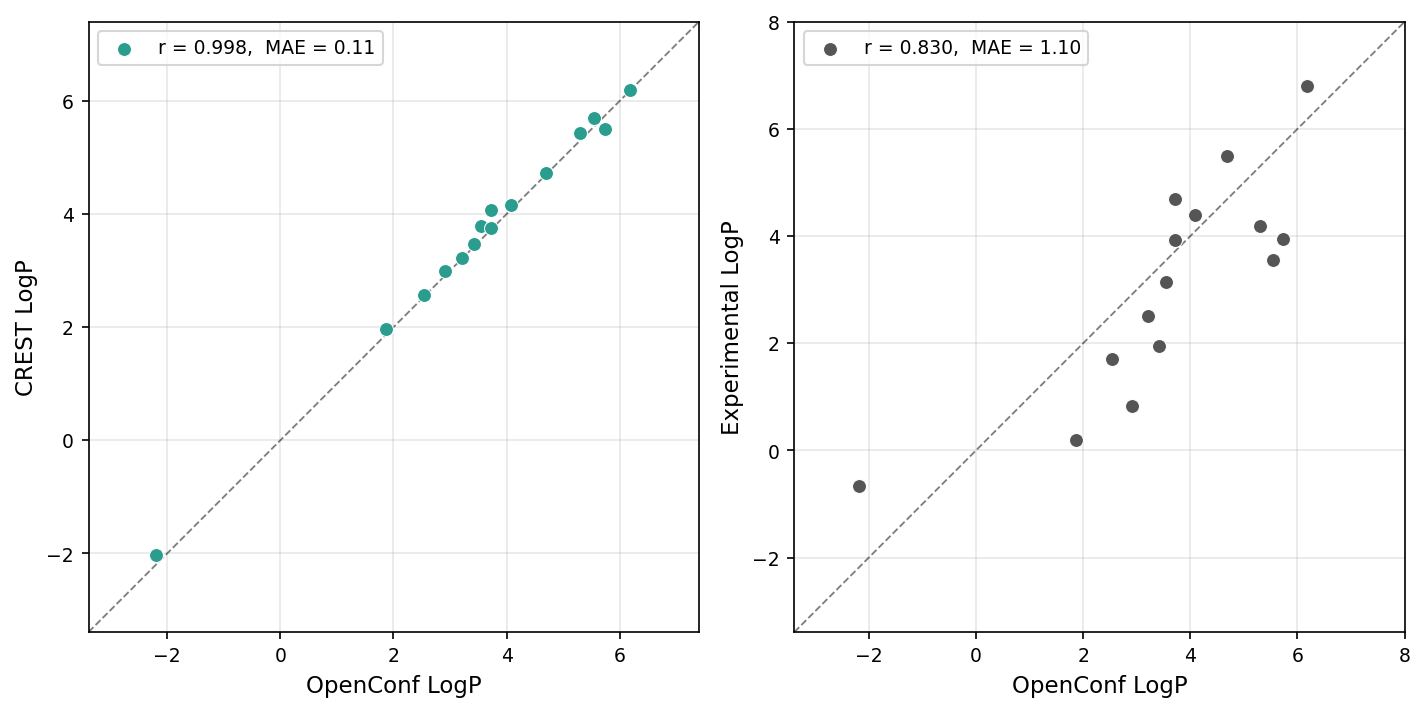
We’ve also looked into using openconf-based ensembles for logP(octanol/water) prediction using Rowan’s solvent-dependent-conformer workflow, following precedent from Novartis on physics-based methods for logP prediction. We found that openconf runs 2 times faster than our previous iMTD-GC-based settings while giving virtually identical results (below, left), producing final logP predictions in line with those reported previously (below, right).
Macrocycles
Macrocycles are challenging for many conformer-search methods. Unlike typical drug-like molecules, macrocyclic conformations are connected by highly coordinated motions of the ring system. While these conformational changes can be described in terms of torsional rotations, they often require many dihedral angles to move simultaneously. To address this challenge, openconf incorporates several macrocycle-specific Monte Carlo moves (as detailed above). These specialized moves substantially improve sampling relative to naïve torsional perturbations and enable openconf to discover many macrocyclic conformations that would otherwise be challenging to discover efficiently.
Even with these enhancements, though, macrocycles remain challenging. To improve macrocycle performance further, openconf automatically expands the energy window and includes an optional low-mode conformational search routine. This technique uses the low-energy vibrational modes of the molecular Hessian to explore new conformational basins—since the lowest modes often describe collective molecular “breathing” distortions, exploring along these modes is a handy way to discover new minima that aren’t easily accessed through few-torsion operations (like most macrocycle moves).
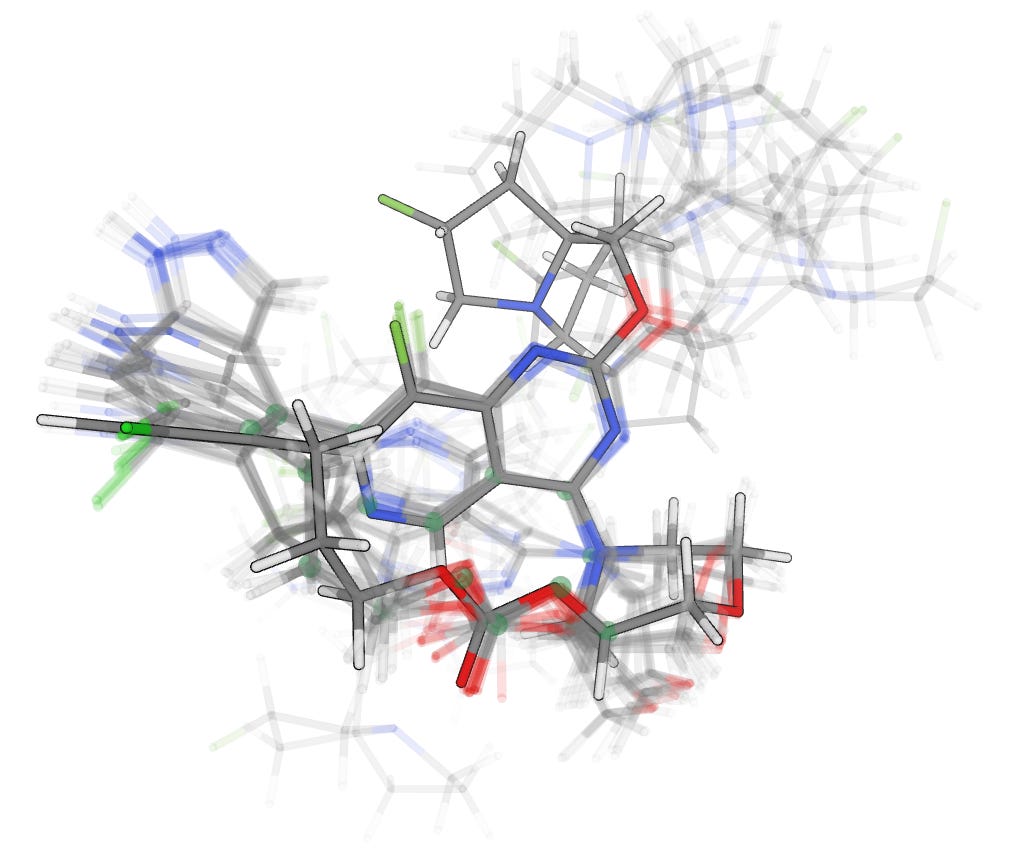
These additions allow openconf to rapidly build diverse macrocyclic conformational ensembles; running a conformer search on the macrocyclic pan-KRAS inhibitor AMG 410 took only 12 seconds and generated 177 unique conformers, 20 of which are overlaid here. While some low-energy conformers differ only in rotation of peripheral groups, larger ring flips and perturbations are also important.
How to Run openconf
It’s straightforward to run openconf on your own projects. Simply run uv pip install openconf and start generating conformers:
from rdkit import Chem
from openconf import generate_conformers
# From SMILES
mol = Chem.MolFromSmiles("CCCCc1ccccc1")
ensemble = generate_conformers(mol)
print(f"Generated {ensemble.n_conformers} conformers")
print(ensemble.summary())
# Save to SDF
ensemble.to_sdf("output.sdf")
# Or XYZ
ensemble.to_xyz("output.xyz")openconf is now the default for Rowan’s conformer-search and solvent-dependent-conformers workflows. Other methods, including ETKDG and iMTD-GC, can still be accessed through the conformer-generator dropdown.
We aim to make openconf an integral part of the open-source scientific ecosystem, not just a Rowan-owned project, and we’re excited to collaborate with other teams on benchmarking and improving the code. If you or someone you know uses an existing conformer-search method, consider trying out openconf and seeing how it compares; if you find any failures, please leave an issue or open a PR on GitHub!
steamroll
We’ve been steadily improving steamroll, our package that generates RDKit molecules and bond orders from XYZ coordinates. We’ve recently integrated code from Jan Jensen and co-workers that works for transition-metal-containing structures and extended their approach to cover lanthanides and actinides too. In practice, steamroll has become much more reliable and no longer freaks out with e.g. propellanes like it used to.
from steamroll.steamroll import SteamrollConversionError, to_rdkit
atomic_numbers: list[float] = ...
coordinates: list[float] = ...
charge: int = 0
try:
rdkit_molecule = to_rdkit(atomic_numbers, coordinates, charge=charge, remove_Hs=True)
except SteamrollConversionError as e:
print("Conversion to RDKit failed!")If you find yourself wanting to convert .xyz files into 2D graph representations, check out steamroll! And, as with openconf, please leave an issue if you find a molecule that can’t be converted. We’ve already found (and fixed) many pathological 2D–3D conversions through Rowan, but more testing is always better for this kind of software.
Chalcedon
We’ve also developed a fast, memory-efficient package for Butina clustering and splitting chemical data after finding existing methods to be slow and impractical for large datasets on previous projects. Our method, Chalcedon, is an order-of-magnitude faster than existing implementations and reformulates the Butina-clustering algorithm to scale linearly in memory instead of quadratically. All performance improvements are made using BLAS through NumPy, making the package dependency-light. We hope to extend this package to include efficient implementations of other clustering and similarity methods as the need arises.
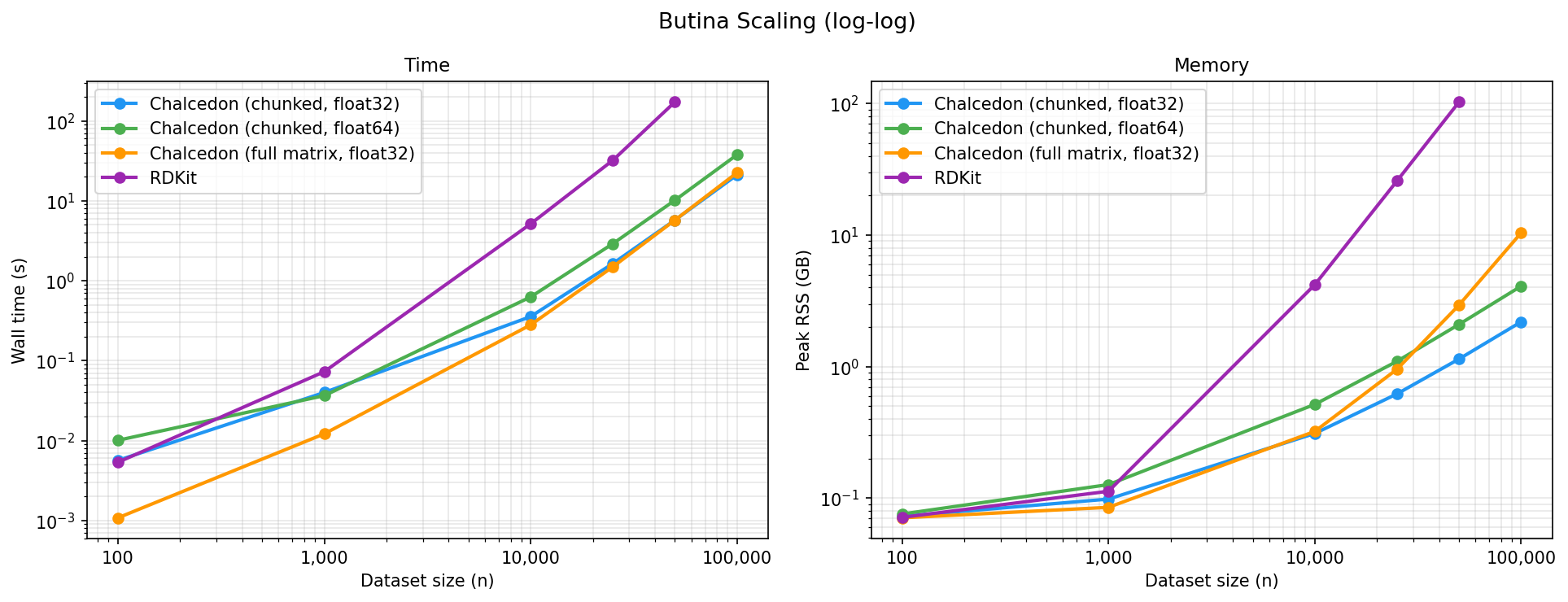
Read more in our blog post announcing Chalcedon from a few weeks ago.
All three of these projects are related to requests we originally made in our call for open-source projects last September. If you’re looking for new open-source chemistry projects to contribute to, consider contributing to one of the projects in that post, making a PR on one of our open projects, or reaching out to our team! We’re always happy to chat about what we think would be useful for the field.
 | A guest post by
|