
Predicting Infrared Spectra and Orb-v3
light and its manifold interactions with matter; why IR spectroscopy is useful; predicting IR spectra through Rowan; Orb-v3



")
This week, we’re happy to share that we’re adding infrared (IR) spectra predictions and Orbital Materials’ new Orb-v3 atomistic simulation model to our platform.
Spectroscopy in Analytical Chemistry
Often, organic chemists are faced with the question: “what molecule is this white powder that I made?” Despite its seeming simplicity, this important question consumes a large amount of lab resources. Chemists frequently rely on the tools of analytical chemistry—including infrared (IR) spectroscopy, thin-layer chromatography (TLC), mass spectrometry (MS), and NMR—to answer questions like this.
Spectroscopic tools, such as IR, Raman, UV-Vis, vibrational circular dichroism (VCD), and electronic circular dichroism (ECD) operate using the same basic mechanism. A scientist prepares a sample and places it in a spectrophotometer, where the machine shines light on the sample (the frequency depending on the type of spectroscopy) and measures the resulting absorption or emission spectrum. The resulting spectrum contains clues as to the nature of the molecule, and it often takes a trained chemist to understand all of the fine details.
Different spectroscopic techniques give information about different molecular behaviors. In IR and Raman spectroscopy, the light excites vibrational motion in the molecules, with each frequency corresponding to a specific motion of the molecule. UV-Vis excites molecules to different electronic states, with the absorption and subsequent emission (fluorescence) providing information about the electronic structure. VCD and ECD measure the difference in absorption between enantiomers, providing insight into stereochemistry by observing the interaction between vibrations (VCD) or electronic structure (ECD) and circularly polarized light.
While all spectroscopic methods can in principle be modeled computationally, some are more amenable to computation than others. Electronic excited states are notoriously challenging for quantum chemistry to get right, making UV-Vis predictions somewhat error-prone. In contrast, infrared and Raman spectroscopy depend only on the ground-state potential-energy surface and can often be computed with very high accuracy.
While IR spectroscopy is no longer employed as a routine part of the organic chemist’s characterization toolkit, there are still plenty of advantages to IR spectroscopy:
The infrared timescale is very short (c. 10-13 s), allowing for very fast processes to be studied by IR. In contrast, NMR spectroscopy is much slower and often can’t give information into e.g. solution-phase conformational ensembles. (See Corin’s blog for more on timescales in chemistry.)
IR spectroscopy doesn’t require samples to be dissolved—you can obtain IR spectra from solids or polymers, which are otherwise almost impossible to characterize.
IR spectrometers are cheap, robust, and reliable, unlike many other spectroscopic instruments. (Ever had to refill the N2 on a Mössbauer spectrometer in the middle of the night?)
And for certain functional groups, like nitriles, allenes, isocyanates, and diazo groups, there’s a certain IR absorbance that’s (1) extremely visible and (2) incredibly diagnostic.
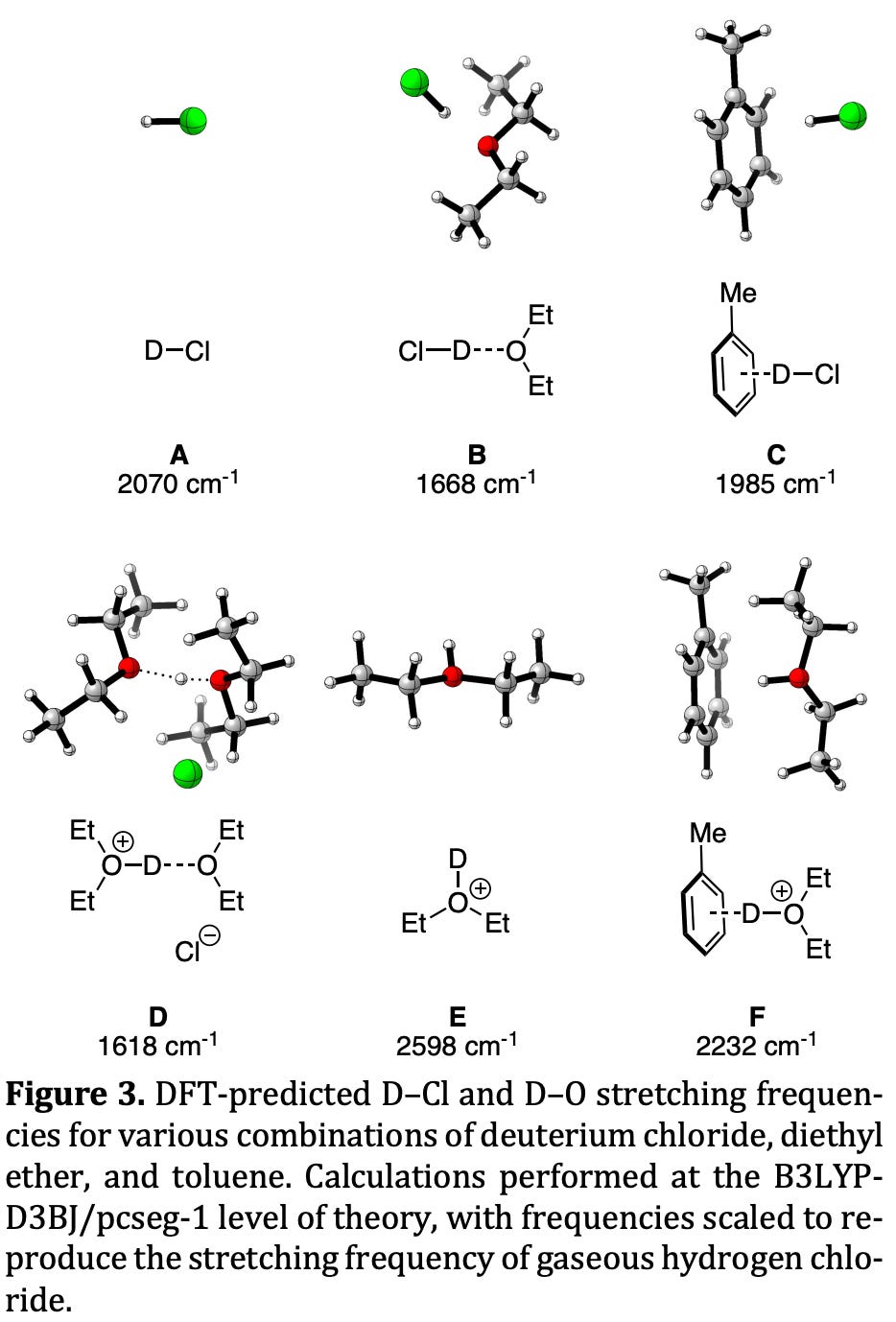
The ability to quickly and accurately predict the IR spectrum of unseen molecules is also very valuable, and allows for IR spectra to be used in tricky structural assignment problems. In graduate school, Corin from our team matched DFT-computed IR spectra to experimental data to determine the solution structure of ethereal hydrochloric acid. The ability to quickly rule out structural hypotheses on the basis of computational data was key to this project:
Infrared Spectra Predictions
You can now use Rowan to predict the IR spectra of molecules computed using DFT and semiemprical methods. To predict a molecule’s IR spectrum, submit a new “Calculation” with the “Frequencies” task selected using an xTB method or a DFT method in Psi4.

The absorption intensity of each frequency is determined by calculating the derivative of the dipole-moment along each mode (typically with CPHF/CPKS). The IR spectra is then created by convolving individual peaks with a Gaussian curve having a full-width-at-half-maximum of 15 cm-1 (as described by Jon Kragskow and Sebastian Dechert).
When the calculation is finished, the resulting spectral prediction will appear in a new “IR” tab.

To animate the largest contributing vibrational mode at a given wavenumber, click on the graph. You can switch between these modes using your left and right arrow keys. To animate a specific vibrational mode and view its contribution to the spectrum, find it in the dropdown box below the spectrum.
Here’s an illustration of a particularly informative IR spectrum, the spectrum of para-cyanobenzaldehyde. You can see sharp peaks corresponding to the aldehyde C=O stretch (1759 cm-1), the nitrile C≡N stretch (highlighted in black, 2386 cm-1), and the aldehyde C–H stretch (2774 cm-1). These values were computed using GFN2-xTB; more accurate values could be computed with DFT.

While we don’t yet support any other form of spectroscopy, we’re always brainstorming about what to do next, and would love input. If you’re interested in using computational tools alongside analytical methods, we’d love to hear from you at contact@rowansci.com.
Orb-v3
Last week, we were excited to be a launch partner for the new Orb-v3 model from Orbital Materials. Orbital Materials is an awesome startup working to use AI models to design new materials and then take those materials to market.
Orb-v3 is the latest in their series of Orb neural-network-potential models. These models simulate systems with all-atom resolution at an accuracy approaching routine density-functional theory, and Orb-v3 can now scale to simulations of 100,000 atoms while performing an energy and force evaluation in under 1 second.
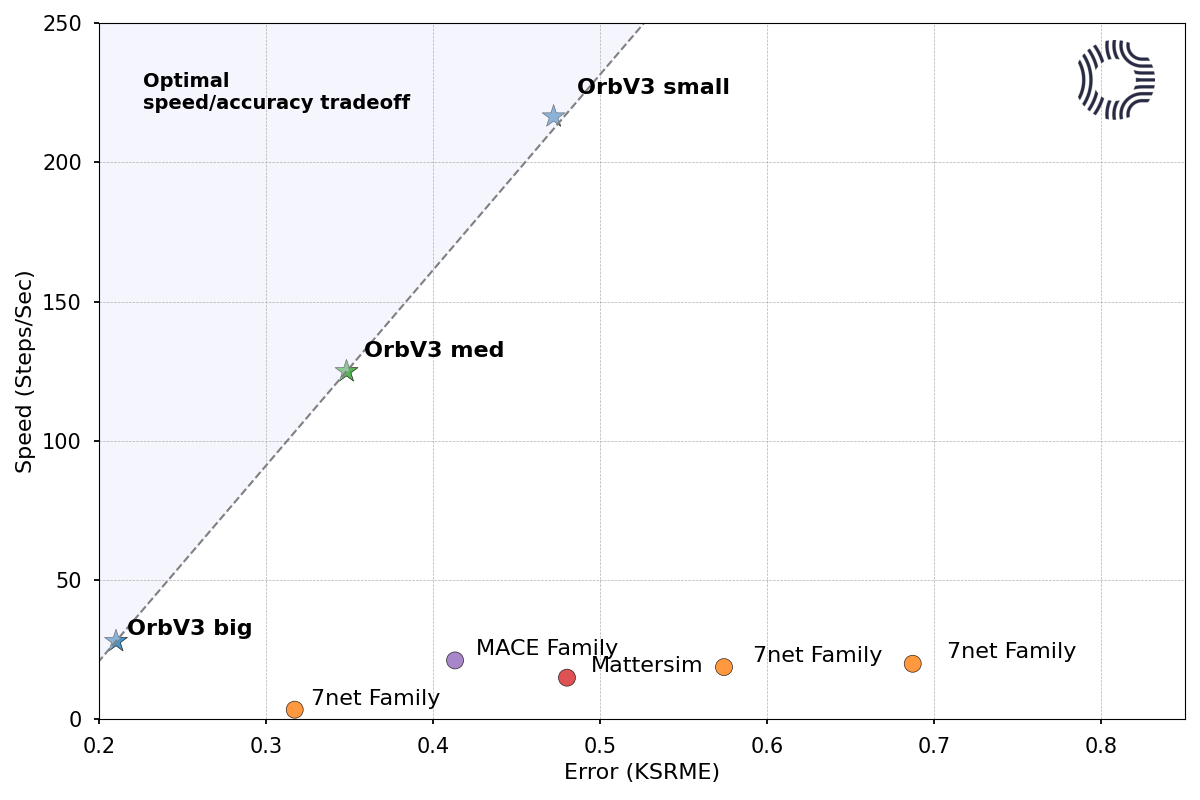
To read more about Orb-v3, see their blog post, the orb-models GitHub, or the preprint. To get started running Orb-v3 on your own structures in seconds, you can use Rowan’s computational platform.