
Projects: Organization, Sharing, and Saving Structures
better organization through projects; saving structures; usage tracking; new conf. search features; second-order SCF; ex. API repo; SMILES imports; a guide to the pKa-perplexed; our inaugural demo day





Today, we’re introducing a new way to organize and interact with computational chemistry through Rowan’s web application alongside a number of smaller features and improvements to existing capabilities.
Stay Organized with Projects
When using a tool regularly, it’s easy to get disorganized. We’ve noticed that users have jobs piling up in their home folders, and sometimes it can take a while to find a particular calculation.
Today, we’re rolling out a new top-level organization entity, which we’re calling “projects.” Each project has its own home folder, so you can start fresh at any point by creating a new project.
You might start a new project: after publishing a paper and beginning a fresh research direction; when beginning work on a new therapeutic target; by assigning each lead compound its own project to keep synthesis, screening, and in vivo preparation neatly contained; or whenever you think it makes sense.
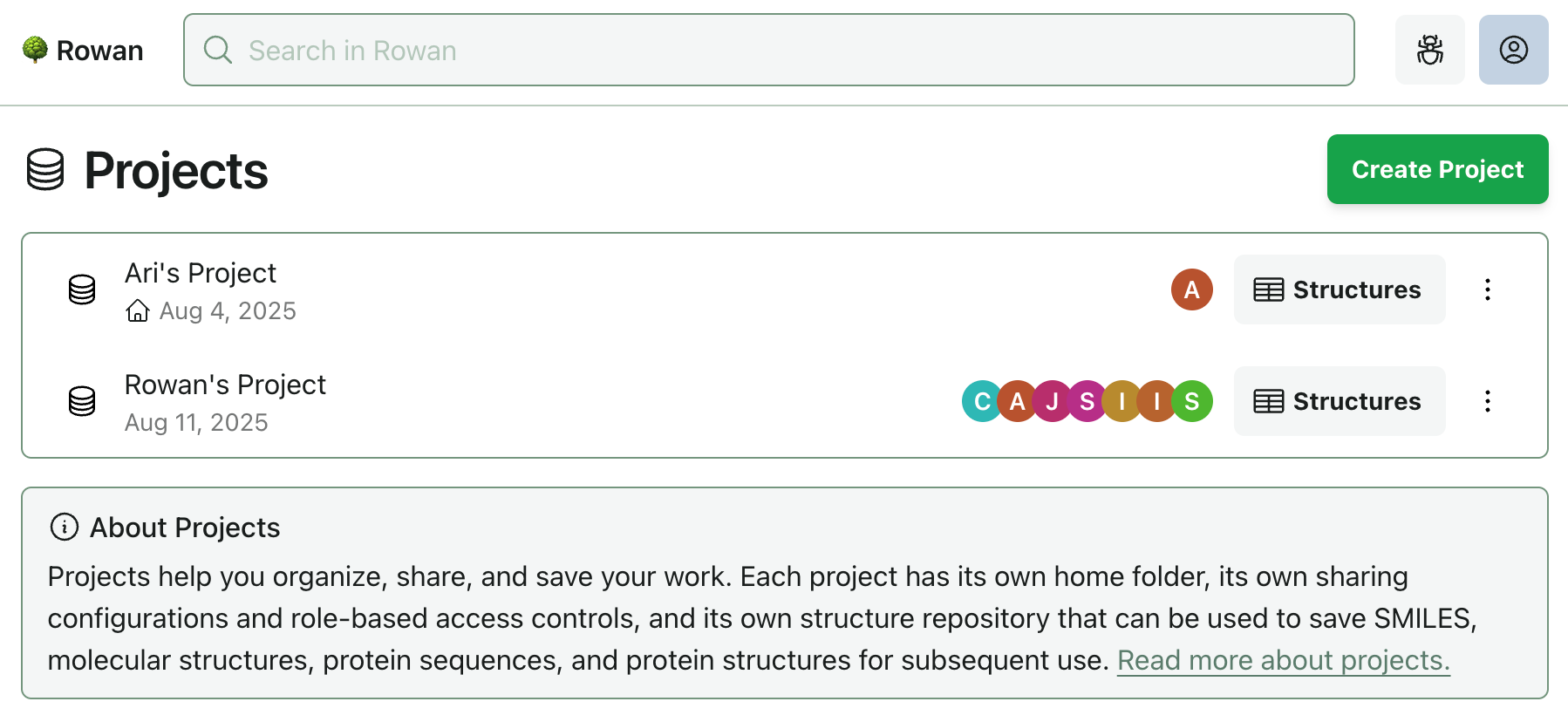
(Additionally, for the times when you really can’t find something, we’ve added a new search interface at labs.rowansci.com/search.)
Managing Sharing
As more teams use Rowan, we've seen the limits of our simple two-tier sharing model (user and organization levels). With this release, we’re giving projects their own sharing configurations and role-based access controls. Each project has one owner, and you can add any number of collaborators from your organization.
When using the API, jobs submitted without a folder_uuid were previously created within a user’s home folder. In the new projects-based paradigm, jobs submitted without a folder_uuid will be created within the home folder of a user’s selected “default” project. Any project you own can be set as your default, and the project you’ve set as “default” cannot be deleted.
Saving Structures
We’ve received a number of requests to save sets of XYZ coordinates and protein sequences inside of Rowan for future use and job submission. With this release, we’re adding project-level structure repositories that let you save SMILES, molecular structures, protein sequences, and protein structures. You can create new project structures at any point, and you can quickly load them into any job submission page for subsequent use.

We’ve got a lot more planned for these “project structures.” We hope that this new way of interacting with compounds inside of Rowan will spark thoughts and feature requests, and we’re excited to hear your ideas. If you’ve got a use case you’d love to see supported, we’d love to hear from you; send us a message at contact@rowansci.com.
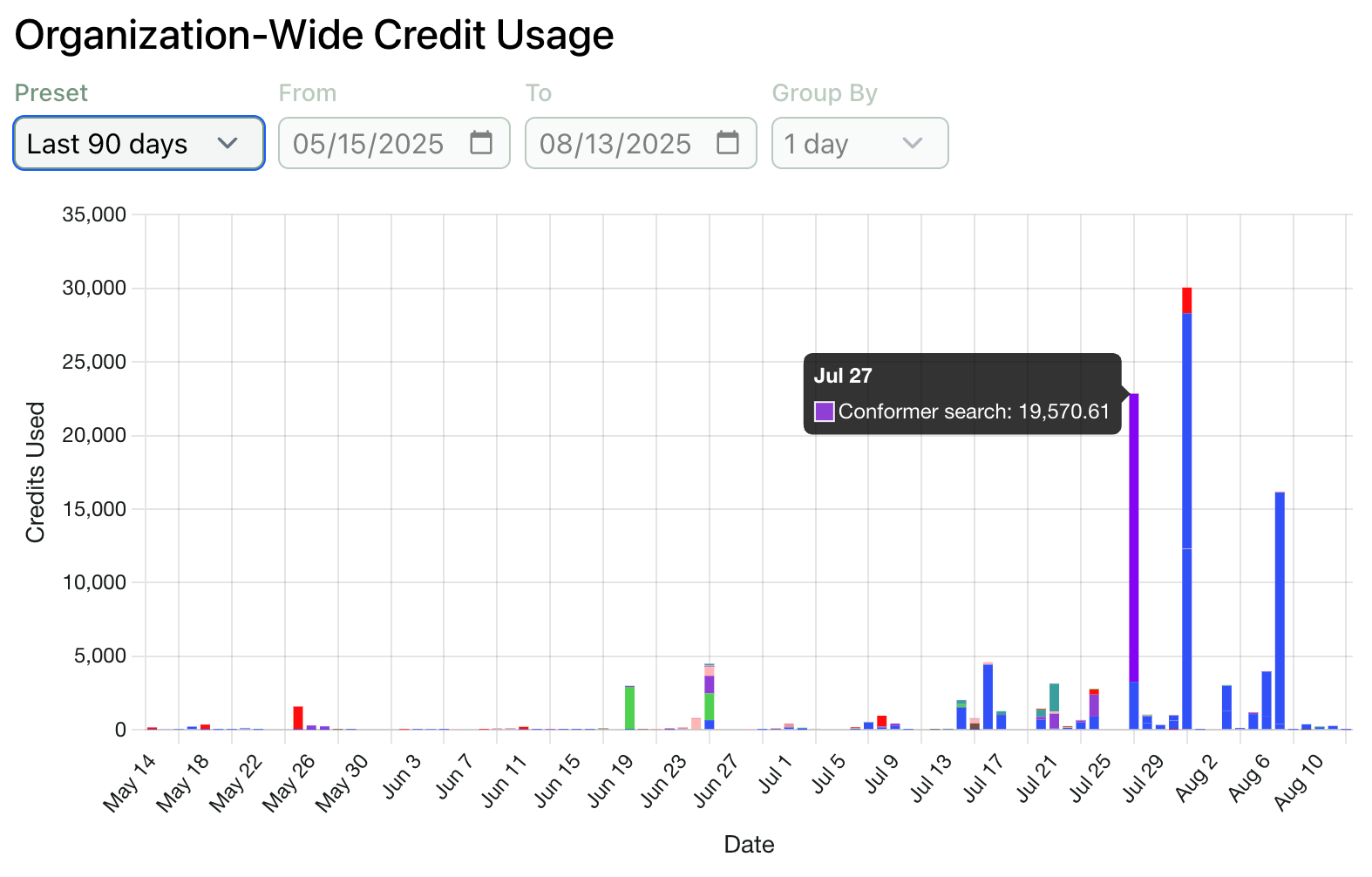
Track Usage Across Your Organization
We’re eager to help administrators understand how Rowan is being used to accelerate their teams. To this end, we’ve added a organization-level version of our credit usage graph, which can be viewed by organization admins on the organization page.
Conformer Search Updates
Two new levels of theory—g-xTB // GFN2-xTB and OMol25's eSEN Conserving Small—are now available as final refinement method options for conformational searches.
We’ve also added a toggle that controls whether or not CREST’s non-covalent interaction (NCI) mode is employed during “Careful” and “Meticulous” conformer searches. NCI mode greatly improves the efficiency and accuracy of running conformer searches on multi-structure complexes: see our post on X.
Improving DFT Reliability With SOSCF
In density-functional theory and related methods, the electronic structure of the molecule is optimized through an iterative self-consistent-field (SCF) process. This optimization is formally a chaos-theory problem and is not guaranteed to converge; while algorithms like DIIS and ADIIS almost always converge in 10–30 steps for simple organic molecules, open-shell systems or metal complexes often prove trickier. In some cases, SCF convergence can become so difficult that getting all the way through a geometry optimization without crashing is almost impossible.
To help address this, we’ve added support for second-order SCF (SOSCF), a slower and more robust SCF-convergence algorithm that almost always works. If SCF convergence failure is detected, Rowan will automatically switch to SOSCF and continue the calculation. This gives users the best of both worlds: routine calculations can run using fast algorithms like DIIS, while the slower and more reliable SOSCF algorithm is used only where necessary. We’ve been trying this out internally and have seen big improvements for various organometallic complexes.
Kickstarting Your API Usage
We’ve tried to make our API as simple as possible to use, and we’re very proud of v2 of our Python API, which is available to all Rowan users for free. However, Python package management is the gift that keeps on giving—see this relevant xkcd comic—and we’ve heard that a number of users have had issues getting rowan-python to work using conda.
To help API users hit the ground running with a working environment, we’ve created a template repository on GitHub to put most of the Python environment setup work on autopilot: https://github.com/rowansci/rowan-sample-env. The repository contains clear, step-by-step instructions that make it easy to run your first calculation through Rowan’s API using modern Python best practices:

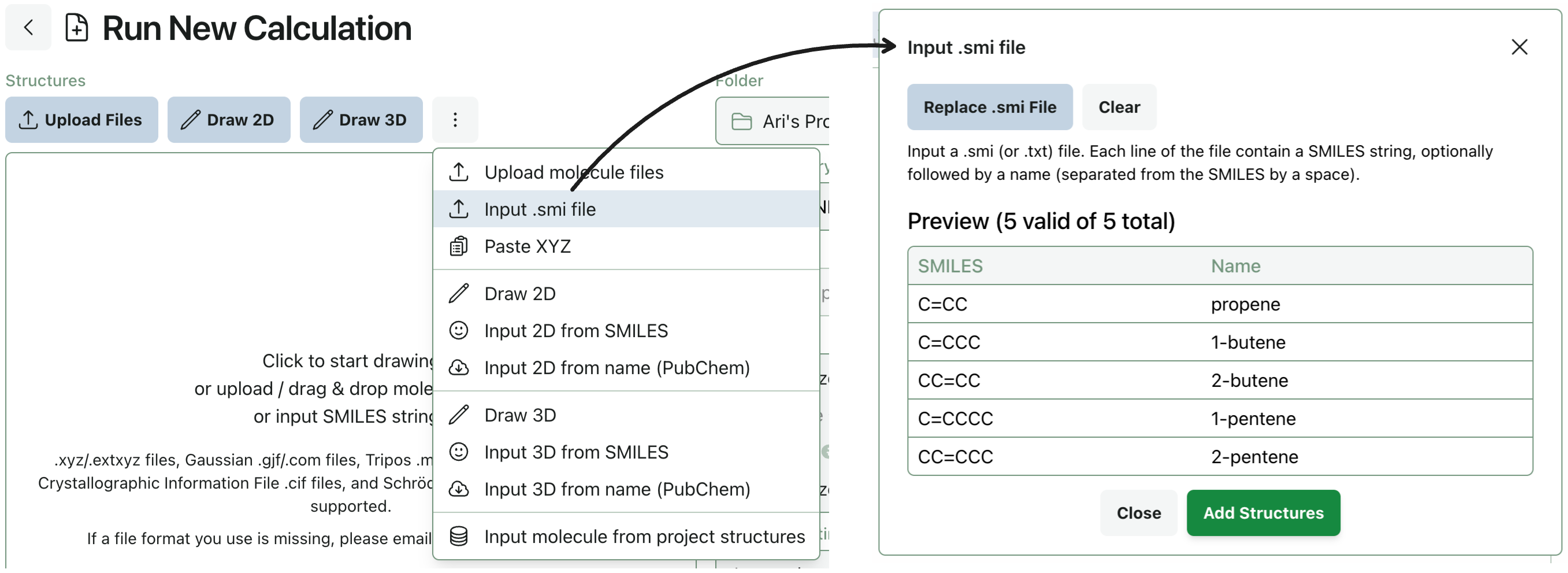
Input Compounds from .smi Files
Some scientists are in the habit of storing collections of SMILES and sometimes names in text files with file ending .smi, .smiles, or simply .txt. You can now import these files into Rowan as either project structures or for immediate submission using a new .smi file utility we’ve built.
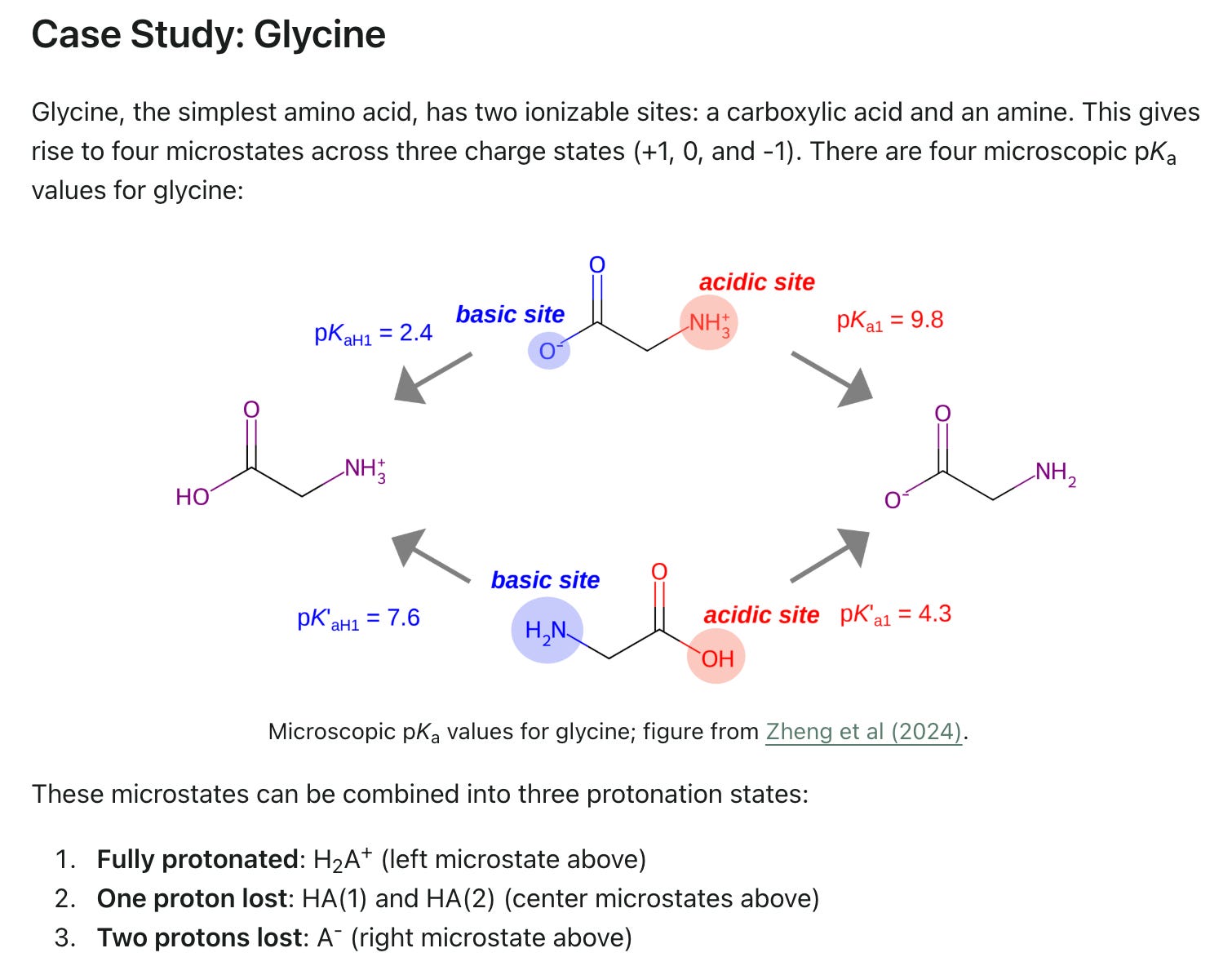
Macroscopic and Microscopic pKa
Rowan now offers both (ML-accelerated) macroscopic and microscopic pKa calculations: unfortunately, the difference between these two paradigms is often neglected or misunderstood. In our latest blog post, we explain the difference between macro-pKa and micro-pKa, give examples of molecules where the two are different, and discuss when one or the other should be used.
Rowan Demo Day
On Wednesday we hosted Rowan's inaugural "Demo Day," where our interns presented what they'd been working on to our team and external guests from industry and academia:
Ishaan Ganti - new approaches for binding-affinity prediction
Vedant Nilabh - ML approaches to conformer generation, plus some thoughts about benchmarks and data in this space
Isaiah Sippel - building modern and interactive educational content through Rowan
Sawyer VanZanten - benchmark results for OMol25-based NNPs for redox potentials and gas-phase electron affinities.
Additionally, Clay Kosonocky gave a special guest presentation on his work running the "Bits to Binders" global protein-design challenge. Corin and Jonathon also got in on the fun, talking about their respective projects on modeling ion-mobility mass spectrometry and TS-finding methods (both of which we hope to release soon on the Rowan platform).
For pictures, check out the discussion on social media (LinkedIn, X).