


Protein–Ligand Co-Folding
folding vs co-folding; free open-source models; running Boltz-1 and Chai-1 through Rowan; decentralized data generation with Macrocosmos




Today, we’re excited to share that we’re launching protein–ligand co-folding on Rowan.
Protein–ligand co-folding is one of the most exciting areas in computer-assisted drug design right now. Previously, the advent of protein-folding algorithms made it possible to go from a protein’s 1D sequence of amino acids to its 3D structure—a task which was previously one of the biggest unsolved challenges in biology. The success of deep-learning-based methods like AlphaFold at this task is probably, to date, the biggest achievement of machine learning in the life sciences.
Co-folding algorithms are the next generation of this technology: in addition to folding proteins in isolation, they can simultaneously generate 3D structures of protein–ligand complexes, based only on 1D representations of the protein and the ligand. And they’re not limited to just single protein–small-molecule complexes, but can also handle other biomolecules, like RNA and DNA, and higher-order complexes.

These capabilities are, quite frankly, mind-blowing. Co-folding algorithms allow scientists to quickly see how changes to a protein’s sequence might impact the structure of downstream biomolecular complexes, a task which used to be virtually impossible without extensive experimental resources. Workflows that used to take months or years of experimental X-ray crystallography or cryo-EM can now be done in just minutes. There’s a reason this area of research won the Nobel Prize last year!
While some protein–ligand co-folding algorithms serve as the underpinnings of a proprietary therapeutics platform (like DragonFold from CHARM or NeuralPlexer from Iambic), new models like Chai-1 and Boltz-1 are free and open-source. Rowan makes it fast and simple to run these models—simply put in your protein sequences and small molecules, select a model, and hit run!

The model generates the barnase–barstar structure shown below in just a few minutes, alongside a few scores indicating the model’s confidence in these predictions. (As always, Rowan employees don’t have access through Rowan to this data; we’re happy to sign NDAs with businesses or discuss single-tenant/on-cloud hosting for enterprise customers.)
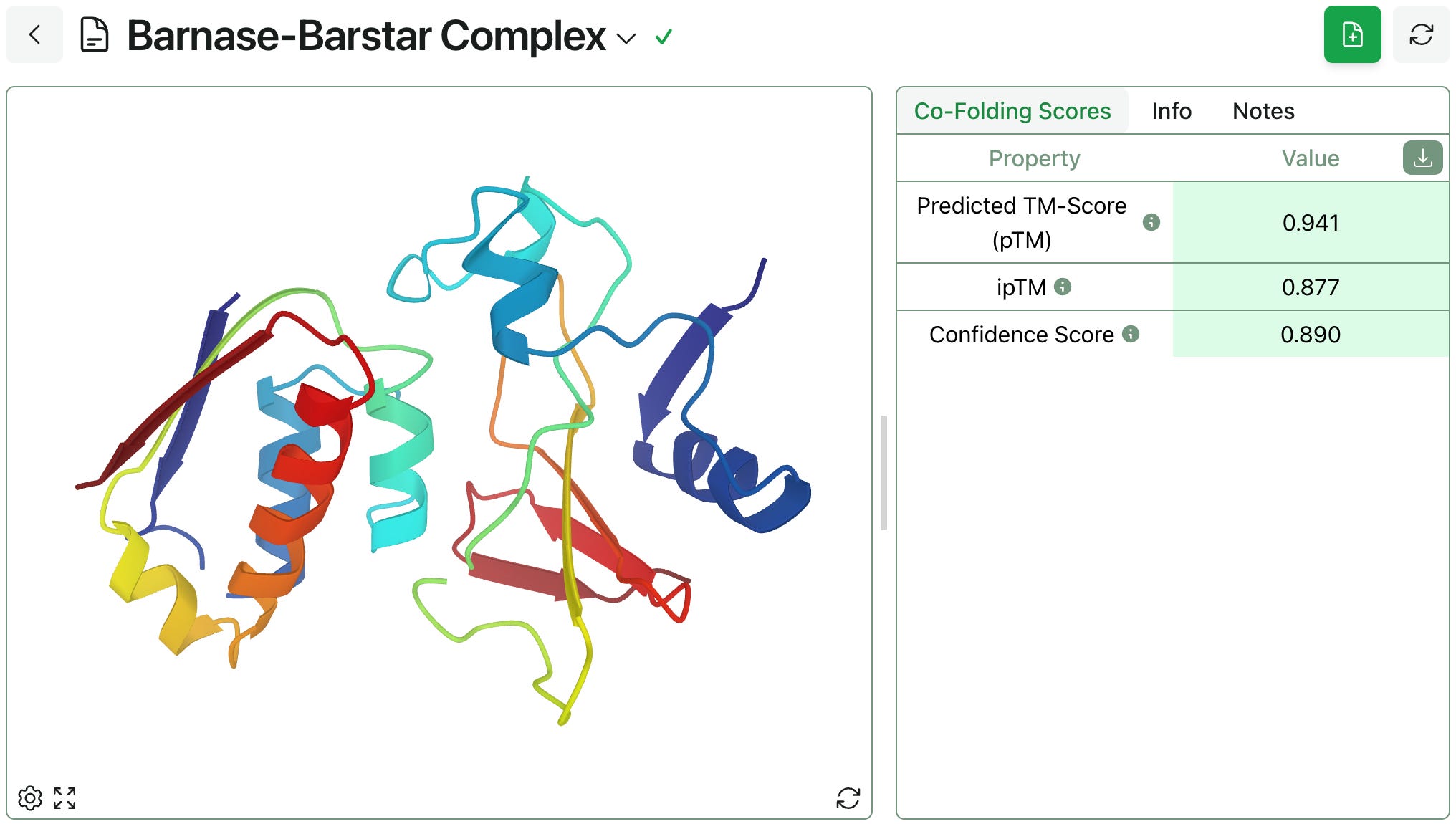
If desired, the predicted structure can be downloaded and used for further analysis.
Our initial implementation of Chai-1r and Boltz-1 isn’t perfect; we don’t yet support restrictions with Chai-1r, we don’t use an multiple-sequence-alignment (MSA) server (which decreases accuracy), and bits of the resubmit and visualization user interface are still a little clunky. We plan to keep iterating and improving on this in the weeks to come.
Our goal is to integrate the best of physics-based and machine-learning methods into a reliable, interoperable, and secure platform for molecular design and simulation. We’re excited to continue expanding into protein modeling and structure-based drug design. If you have suggestions for additional functionality in this area, please let us know!
Partnering with Macrocosmos
Last week, we released Egret-1, a family of neural network potentials for biomolecular simulation. These models were trained on millions of high-accuracy density–functional theory (DFT) calculations, and we believe improved dataset scale and quality will contribute to improvements in accuracy and generality in the next generation of models. To that end, we’re partnering with Macrocosmos to leverage the decentralized computing power of Bittensor Subnet 25 - Mainframe to generate DFT data. See our blog post for more details.