
Protein–Ligand Docking
critical contemplation of the merits and demerits of docking; unphysical poses and how to find them; machine learning offers a pragmatic solution; running docking on Rowan; future protein aspirations




Today, we’re excited to announce that we’re adding protein–ligand docking to Rowan.
Docking: Friend or Foe?
Protein–ligand docking is the archetypal computer-assisted drug design workflow. Docking directly addresses the key questions of “how will this compound bind to my target” and “will it be any good,” making it immediately relevant to almost every campaign, and for this reason docking is many people’s first exposure to computational chemistry (as it was for Corin on our team). When people want to visualize the field of computer-assisted drug design, they usually highlight docking:

Unfortunately, docking also doesn’t work very well. In most cases, protein–ligand binding is poorly described by a single conformation, meaning that trying to quantitatively compare docking scores to binding affinities is an exercise in futility. In a recent post, Avery Sader states this clearly (emphasis original):
Docking can be useful for generating hypotheses of binding modes. But here’s the thing: docking cannot predict binding affinities… Binding affinity depends on the entire free energy landscape, not just a single docking pose. Protein dynamics and conformational entropy are huge factors. Kinetics and residence time also influence binding affinity, but are not accounted for in docking.
The failure of docking to reproduce experimental IC50 values has been documented many times (e.g.), so why bother with docking at all? Here’s why we think docking is useful, and what we hope our users get from this new workflow:
Docking lets you test binding poses. A good docking score by no means implies that the molecule will have a high binding affinity—but a molecule that cannot fit into the binding pocket is unlikely to have a high binding affinity. Docking allows us to quickly test the effect of structural modifications and avoid synthesizing & testing compounds that are virtually guaranteed to be inactive.
Docking gives us poses for further analysis. Even if docking scores themselves aren’t particularly accurate, the geometries generated by docking are often a good starting point. These geometries can be used for a whole host of more advanced methods: molecular dynamics, MM-GBSA or MM-PBSA, free energy methods like FEP or TI, QM- or ML-based rescoring functions, and more methods that are being developed every day. Docking is the fastest and most practical way to generate these poses.
For some applications, docking is the only structure-based technique that’s fast enough. As chemical libraries get larger and larger, searching through them requires sophisticated techniques and lightning-fast binding-affinity prediction methods. The state-of-the-art solution here remains docking, in conjunction with a sophisticated ML- or cheminformatics-based orchestrator that chooses which compounds to dock next (see inter alia 1, 2, 3, 4).
Accounting for Strain in Docking
As discussed above, protein–ligand docking suffers from a few pathologies that often cause docking scores to differ substantially from experimental binding affinities. Some of these reasons are difficult to handle without resorting to top-shelf methods like FEP: ligand conformational motion, pose flexibility, explicit interactions with water, and so on and so forth. But others are more prosaic. Docking algorithms are prone to generating strange and unrealistic ligand poses which bind with ultra-high affinity—these false positives can mislead scientists and waste time and money.
These faulty poses can be identified by computing the pose’s strain relative to the global minimum. Careful studies of experimental protein–ligand complexes has shown that the total strain on the ligand (Ebound − Emin) rarely exceeds 5 kcal/mol when computed by DFT (1, 2, 3), and Brian Shoichet and co-workers have shown that filtering out high-energy poses can significantly improve the success rate in large-library docking. Unfortunately, computing per-pose strains with DFT is far too slow to apply to large libraries, and so Shoichet and co-workers had to use pre-computed libraries of four-atom torsional profiles to estimate the total strain. This is an ingenious solution, but almost certainly misses important higher-order effects like intramolecular hydrogen bonding.
Since neural network potentials (NNPs) have recently emerged as an appealing low-cost alternative to DFT for ranking conformer energies (and benchmark well at this task), we envisioned that we could use NNPs to generate strain energies for the entire ligand. Following Butler and co-workers, we optimize each pose with harmonic constraints tethering each atom to the original structure to prevent bizarrely high strain energies that result from minor differences in bond lengths or angles at different levels of theory. (OpenEye’s Freeform does something similar.) We then compute single-point energies at the AIMNet2/CPCMX(water) level of theory, and compare these to the energy of the lowest-energy conformer generated by our initial conformer search.
Our overall docking workflow works like this:
Screen conformers with GFN2-xTB to remove duplicates and high-energy conformers. Then, reoptimize each geometry with AIMNet2.
Compute the energy of each conformer with AIMNet2, adding a ∆Esolv correction from CPCMX(water) at the GFN2-xTB level of theory.
Use AutoDock Vina to generate docked poses and scores for each conformer using the Vinardo scoring function.
Optimize each docked pose with AIMNet2, employing a 5 kcal⋅mol-1⋅Å-2 harmonic constraint on the position of all heavy atoms. This prevents the pose from dramatically changing, while still allowing bond lengths and angles to relax to reasonable values.
Compute the energy of each pose with AIMNet2, adding a ∆Esolv correction from CPCMX(water) at the GFN2-xTB level of theory.
Return per-pose docking scores and strain energies.
We anticipate that these additional steps—generating a high-quality conformer ensemble and correcting for ligand strain—will noticeably increase the quality of results from docking. This doesn’t “fix” docking, but should help to remove some of the more pernicious pitfalls. Since these extra steps take a bit more time, this docking workflow is probably better suited to hit-to-lead/lead optimization than to ultra-high-throughput virtual screening.

Running Docking Through Rowan
To start a new docking run and accompanying ligand strain calculations in Rowan, start by inputting a protein either by entering its PDB ID or by uploading a .pdb file.

Once you’ve uploaded a protein, use Rowan’s protein tools to remove any duplicate chains and/or save a pocket from a bound crystal structure before “sanitizing” the protein. The sanitization process cleans up any artifacts from the crystal structure and removes non-polymer and solvent molecules, preparing the protein for docking.

On the submit page, you can choose to use the crystal structure's selected pocket, perform a blind dock, or manually define a custom pocket.

After your docking run completes, docking scores will be displayed next to our new interactive protein viewer. Any poses with high ligand-strain energies are automatically flagged.
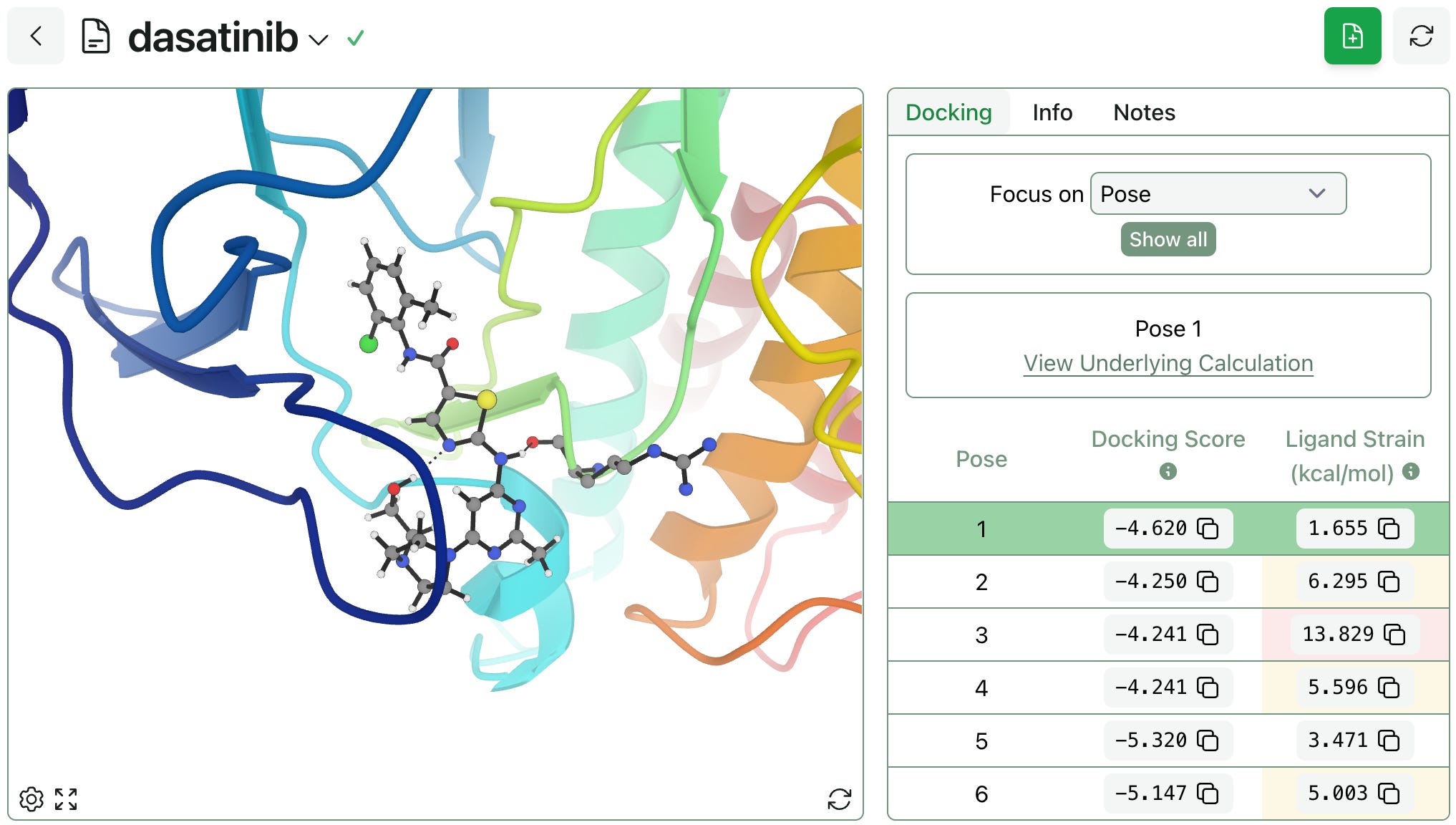
The viewer will focus on your selected docked pose by default. This behavior can be adjusted to focus on the pocket or the entire protein instead, or can be temporarily paused.
In the viewer, residues that interact with your docked ligand will be shown in all-atom view. Through the settings menu (gear icon at the bottom left of the viewer), you can choose to display nearby non-interacting residues and toggle hydrogen atom visibility.
To help you compare results, you can also display all docked poses simultaneously on the protein, providing a comprehensive visual overview of the workflow's output.

Moving Into Proteins
Now that we’ve broken the ice by adding our first protein capabilities to Rowan, we’re excited to further expand our protein-focused capabilities. Over the coming months, we plan to explore protein input tools like sequence-to-structure ML models (AlphaFold 2), additional docking methods, post-docking corrections, and more advanced protein–ligand workflows.
If you're working in structure-based drug design, we'd love to hear what would be most helpful for your team! Reach out to us at contact@rowansci.com.
yep. docking programs give scores that look suspiciously like kcal/mol, but don’t include units, so as not to tempt the wicked.
..several benchmark datasets out there Merck FEP and plinder come to mind. There is this “docking power, ranking power, screening power” thing (I think from Renxiao Wang pdbbind guy https://pubmed.ncbi.nlm.nih.gov/19358517/).
might also mention gnina… https://github.com/gnina/gnina
I have tried making custom scores using docking score and a strain estimate (much less careful - just delta E (mmff) between docked pose and lowest energy conf) - fitting a model to potency/affinity data, using those things as “features”…. Results: meh
Congratulations! One problem I've run into when using NNPs to estimate strain post-docking was the preservation of stereochemistry after the docking run. Most of the time the docking tool is dropping hydrogens, leaving one with the task of adding them back. Currently, I am using explicit stereo-tags in rdkit and an energy minimisation with fixed heavy atom positions. I wonder wether there is a more elegant solution that's worked for you, if you mind sharing!