
Rowan Goes Periodic
periodic vs molecular; why periodic matters; tblite; open materials 2024 on GPU; more in the future




In computational chemistry, there are two types of calculations: molecular and periodic. We use these to simplify the world down to tractable calculations, instead of computing all possible interactions between all atoms.
Molecular calculations are exactly what they sound like—isolated molecules or groups of molecules surrounded by a vacuum (or a dielectric field). This is good for studying small molecules, clusters, or even larger biomolecules.
However, most physically relevant materials are so large as to be effectively infinite relative to the molecular scale. Cutting out a chunk of these materials and modeling them with molecular calculation introduces significant edge effects. To solve this problem, we can use periodic calculations. Materials can be modeled using a single unit cell, where the molecule or group of molecules “sees” itself tiled infinitely in all dimensions. For instance, here’s a snapshot of seven periodic boxes of solid naphthalene. Where the molecule extends out of one side of the cell, it comes back on the other side:

Periodic calculations are great for a lot of tasks. To name just a few:
Most crystals are inherently periodic, and periodic calculations allow direct study of the geometries, densities, and relative energies of different crystal structures. This is important in the pharmaceutical chemistry, where understanding the relative energetics of different crystal forms is crucial for ensuring reproducible and stable API crystallization. (Predicting crystal structures with DFT is also expensive. A recent crystal-structure-prediction competition saw 28 teams run over 46 million CPU-hours of computations!)
Liquids have significant edge effects, so periodic calculations allow descriptions of liquid densities and dynamics. This is not only good for aqueous systems, like proteins and other biomolecules, but also solvent mixtures, battery electrolytes, melts, ionic liquids, and so on and so forth.
Many reactions and processes are better described through periodic calculations. Heterogeneous catalysts are best modeled as an infinitely repeating surface on which a reaction takes place. Similarly, reactions in explicit solvent, phase transitions, or reactive polymerizations all require calculations that are devoid of edge effects.
Unfortunately, most tools in the computational chemistry stack are intended for either periodic systems or molecular systems, not both. As a result, two largely separate ecosystems of researchers, software, and publications have emerged, and it’s not uncommon for a given scientist to have extensive experience in molecular systems while never having run a periodic calculation. We’re really excited to launch periodic calculations today, which will help bring this excellent suite of techniques into Rowan’s platform and make it simpler and faster for scientists to innovate across disciplines.
Periodic Calculations in Rowan
Periodic calculations can be submitted on Rowan just like molecular calculations. If you upload or draw a periodic system, Rowan will populate appropriate level of theory presets. You can also choose whether or not to optimize the cell size during optimizations.
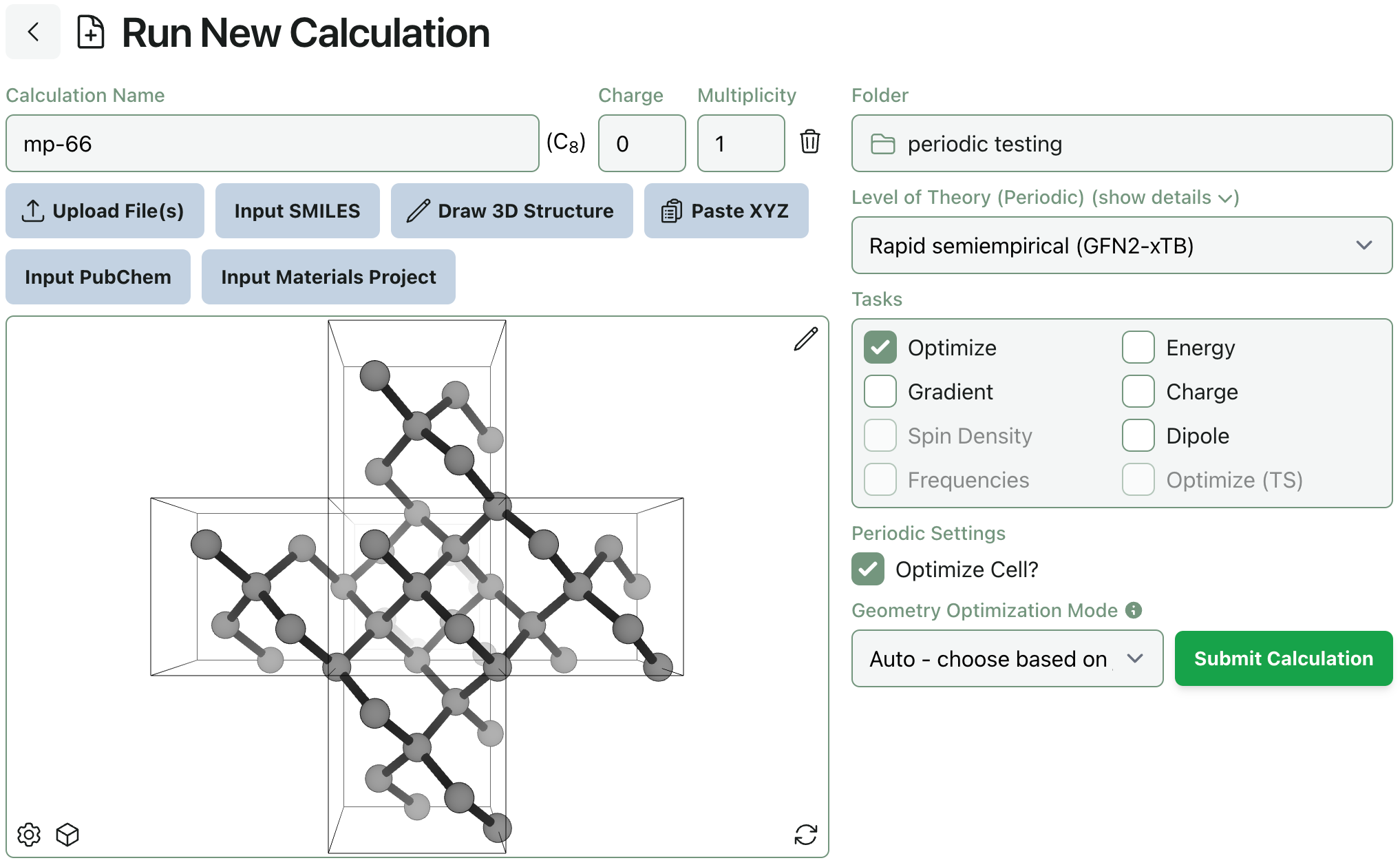
To make drawing periodic systems easier, we’ve added an “Edit cell” tool to the molecule editor. With this tool selected, you can quickly add or remove a cell, center your system inside a cell, edit the cell’s periodic boundary conditions, and directly modify the cell’s dimensions.

We’ve also added a new set of view settings to periodic calculations, so you can quickly toggle whether or not adjacent cells are shown.

To make it easy to add existing materials, we’re launching with an API to the Materials Project database. We plan to add an API for the Cambridge Crystallographic Data Centre soon, making it possible to load any published small-molecule crystal structure with the click of a button.
We’re launching with two periodic engines: xTB and Open Materials 24.
xTB
Users of Rowan will already be familiar with the xTB semiempirical methods from Stefan Grimme and co-workers at the University of Bonn. xTB methods (as implemented in the TBLite library) perform well for the geometries of periodic systems—they’re not as accurate as high-level DFT calculations, but are quick and almost always very reasonable. (For instance, here’s a paper showing that xTB performs well on metal–organic frameworks.)
Open Materials 2024
A week ago, Meta’s FAIR-chem launched a massive new dataset and model focused on inorganic materials, which they’re calling Open Materials 2024 (OMat24 for short). The authors generate a dataset of 118 million periodic DFT calculations using sophisticated sampling techniques, focusing on non-equilibrium structures, and train equivariant neural network potentials using the EquiformerV2 architecture plus non-equilibrium denoising to reproduce energies, forces, and stress over this dataset.
The resulting OMat24 models are state-of-the-art on a variety of test sets, largely because they’re so massive—the largest model has 150 million parameters, which is small compared to large language models like GPT-4 but much larger than previous neural network potentials. Here, for instance, is a screenshot of the Matbench Discovery leaderboard, where the 87M-parameter OMat24 model (“eqV2”) is top in almost every category.

We’re thrilled to be launching OMat24 models for subscribers on Rowan today! Because of the size and complexity of these models, we’ve put a decent amount of work into optimizing their inference: we’ve added GPUs to our inference stack, making it quick and easy users to run large ML-based models on state-of-the-art hardware. (Moving forward, we plan to run more ML-based calculations on GPUs for subscribers.)
This is our first step into periodic calculations, but there’s a lot more we’re excited to do. One notable absence in this release is DFT—we don’t currently offer a periodic DFT engine, although we’ll be happy to change that in the future. Reach out to our team at contact@rowansci.com if you’d like to run periodic DFT, and we’d be happy to discuss more! We’re also hard at work planning workflows for common periodic use cases, and would love to hear about what would be most useful: periodic transition-state optimization is high on our list, as is molecular dynamics and related calculations. We look forward to hearing about what we can be doing better!
 |
|