
Rowan's Beta Launch
Five months later, here we are.


We’re excited to announce Rowan’s first product, a cloud platform for computational chemistry!
Over the past 18 months, we’ve been talking to scientists working in drug discovery about how they use quantum chemistry in their research. (We summarized some of what we’ve learned in a previous post.) Several important themes emerged from these conversations:
Some scientists run quantum chemical calculations frequently and find them very useful, but are often frustrated by the limitations of their tools.
Others run high-level calculations only when they absolutely have to, because the calculations require too much time and effort to run.
And almost everyone commented on how frustrating quantum chemistry can be: manually generating input files and allocating computer time is fine when you’re running a handful of jobs, but doesn’t scale to the pace and throughput of industry research. If the computations take more time than the experiments would, what’s the point?
Our product is directly aimed at addressing these problems. Rowan allows you to submit, run, view, and analyze your calculations all through the same easy-to-use web interface.
Submitting calculations to Rowan is as simple as uploading a list of files (or SMILES strings) and selecting what task you want to do. With Rowan, your jobs won’t sit in a queue: we allocate a new computer for each job as soon as you hit submit. And jobs can be also submitted and analyzed entirely in Python, so that experienced users can save time by automating their workflows.
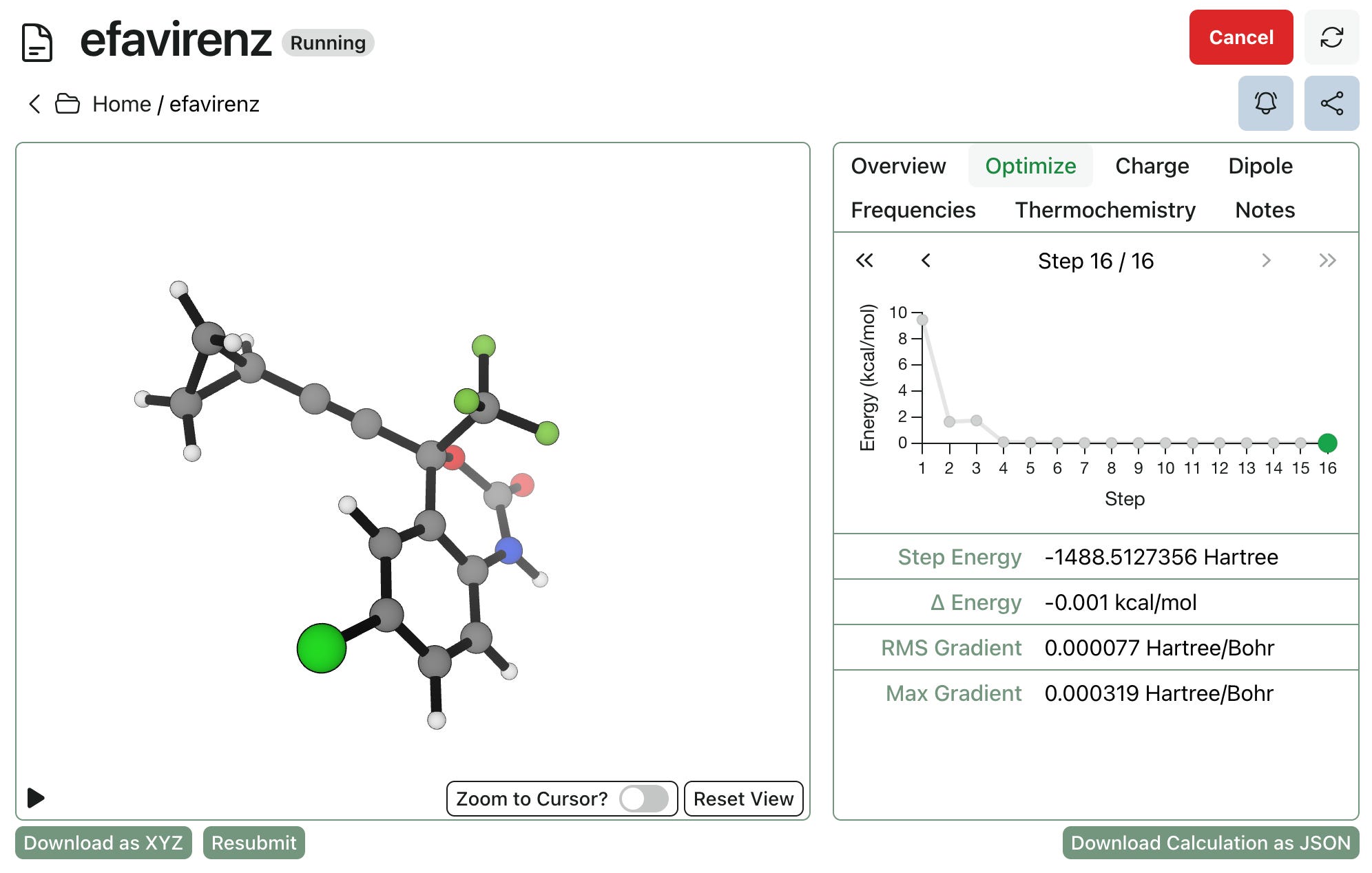
Jobs run through Rowan can be analyzed quickly and easily: Rowan generates publication-quality figures by default, and makes it easy to view and export data into other formats. And results can also be directly shared with collaborators, making it simple for colleagues without computational expertise to view and understand the simulations. (Here’s a job I recently ran, for instance.)
There’s no initial license fee with Rowan: instead, we simply bill you based on the computer time used to run your jobs. So if you’re interested in trying out what we’ve built, go to labs.rowansci.com and sign up for an account—the first 500 minutes are free! (If you’re curious but don’t have any idea how to use this software, don’t worry: in the next few months, we’re planning to demonstrate lots of ways that Rowan can be used to quickly answer important questions in drug design.)
What we’re releasing today is just the beginning: we know there’s a lot of features we don’t yet have, and we’re working hard to fix that. The core scientific codebase is only about 4500 lines of Python, so we’re able to ship quickly—and we have a lot of features in the pipeline, so you can expect Rowan to get noticeably faster, smarter, and easier to use in the coming weeks and months.
If you work in drug discovery and think Rowan might be useful for you, we’d love to chat! Send us an email and we’ll figure out if we can help you with your scientific problems. (And if you’re interested in working with us to increase scientific productivity and shape the future of molecular simulation, let us know by responding to this email.)
Amazing
This looks awesome!! Nice work, you guys!