
Solvent-Dependent Conformer Search
a good conformer is hard to find; clustering and the ReSCoSS workflow; Rowan's implementation, with some expert help; a demonstration on maraviroc


")
Finding the relevant and accessible solution-phase conformers of a molecule is key to accurate downstream simulation. Unfortunately, many molecules are flexible. The number of conformers grows exponentially with the number of hindered rotors, meaning that even many moderately complex drugs have far more conformers than can be systematically enumerated for a reasonable cost.
Even in cases where the number of conformers is “only” a few thousands or tens of thousands, identifying the best low-energy conformations is often non-trivial. Accurate ranking of conformers typically requires energies from dispersion-corrected density-functional theory (DFT), correct handling of solvent effects, and vibrational corrections to give an estimate of the free energy. Because these steps are too expensive to routinely run on thousands of conformers, pre-screening and filtering is typically run with lower levels of theory to reduce the size of the ensemble to a more manageable level—however, these cheaper filtering steps run the risk of incorrectly excluding relevant conformers from the final set.
At Rowan, we try to build practical tools that drive real impact on important scientific problems—accordingly, we’re always quite interested in the reports that come from internal tool-development teams in pharmaceutical companies, since these teams often identify pragmatic and useful solutions to the same problems we’re trying to solve. We were excited to find a 2020 paper from Novartis that reports ReSCoSS, a new workflow to increase efficiency of conformer filtering and ranking through a creative solvent-dependent clustering approach (more on how this works below).
Over the past few months, we’ve had the opportunity to work with Rainer Wilcken, one of the ReSCoSS developers, to adapt this same logic into Rowan. The result is our new solvent-dependent conformer search workflow, now available to all subscribing users. In this post we’ll describe (1) how ReSCoSS works, (2) how we’ve adapted this to Rowan using modern low-cost methods, and (3) why we think this is a useful addition to the modern drug-discovery toolkit.
ReSCoSS and Rowan’s Solvent-Dependent Conformer-Search Workflow
ReSCoSS stands for “Relevant Solution Conformer Sampling and Selection,” and the goal of ReSCoSS is to quickly identify potentially solution-relevant conformers from a large set of generated conformers.
The core insight behind ReSCoSS is that early conformer filtering often fails because inaccuracies in the ranking method will prematurely discard entire classes of conformers. For instance, a forcefield or a low-quality implicit-solvent method might overstabilize the “folded” conformers of a flexible molecule and erroneously discard all the “unfolded” conformers. Even if the final ranking method gets the energies right, it’s too late!
ReSCoSS addresses this by intentionally maintaining the diversity of the conformer ensemble during all filtering steps. Conformers are clustered based on their 3D properties, and conformers from every cluster are kept, even if they’re not globally lowest in energy. This means that the final conformer set always contains substantial 3D diversity, making the workflow more resilient to imperfect energies early on.
Here’s how this works in practice:
Conformer generation. Conformers are generated using any desired input method. After some testing, we found that iMTD-GC with GFN-FF through CREST worked well if we use a fairly high energy window (by default, 30 kcal/mol). As usual, we deduplicate conformers with PRISM Pruner.
Descriptor calculation. Descriptors have to be chosen that allow for conformers to be clustered based on their physical properties. In ReSCoSS, these descriptors come from COSMO-RS; in Rowan, we found that simpler descriptors like solvent-accessible surface area, polar surface area, radius of gyration, and so on were sufficient to let us distinguish different conformer families with good accuracy.
Clustering and selecting. With descriptors in hand, the molecular conformers are now clustered using k-means clustering. From each of the k clusters, the energy of each conformer is computed and the lowest-energy N conformers are selected. (By default, Rowan uses k=5 and N=3.)
To make the procedure even more robust, the energy-based selection is done using energies in many different solvents. ReSCoSS uses ten solvents with different polarity and hydrogen-bonding parameters; by default, Rowan uses hexane, octanol, chloroform, dimethylsulfoxide, and water. For each solvent, the energy of all conformers is computed using GFN2-xTB and the ALPB solvent model, and then the lowest N conformers are selected.
Since up to 5 * 3 = 15 conformers can be selected from each solvent, the maximum number of conformers that can be returned is 15 conformers/solvent * 5 solvents = 75 conformers. In practice, though, it’s common for different solvents to agree on the lowest-energy conformer from each cluster, making 20–30 output conformers more typical.Final optimization and ranking. The small final conformer set is optimized using GFN2-xTB/ALPB(water) to get final optimized geometries, and then the per-conformer free energy is determined using g-xTB single-point energies, GFN2-xTB vibrational free-energy corrections from single-point Hessian calculations, and CPCM-X solvent free energies in each solvent.
The final output is a list of conformers and associated free-energy values in each of the selected solvents. This can be useful in several different ways:
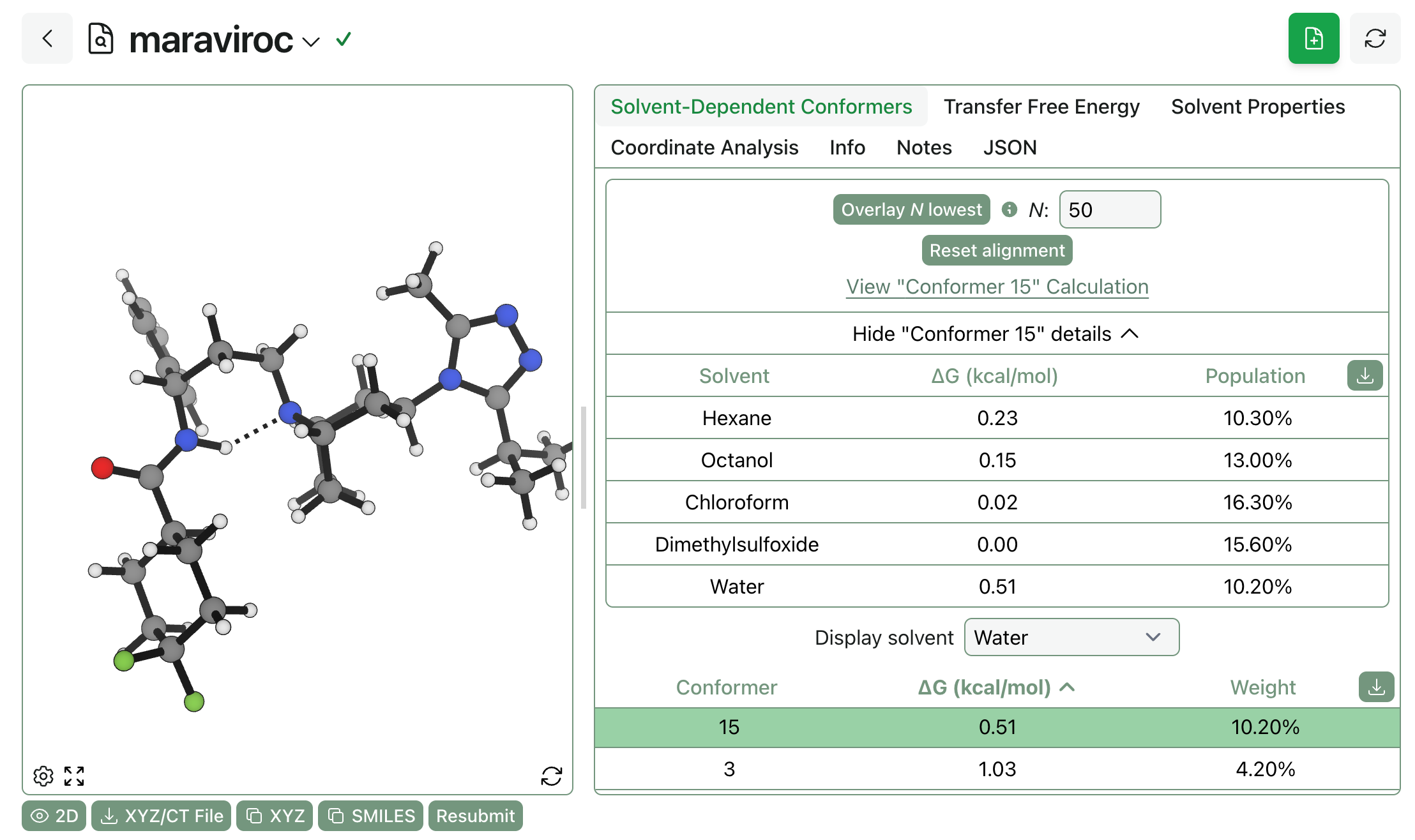
It’s simple to assess the difference between the population of different conformers in different solvents. Just compare the Boltzmann weights of a given conformer across solvents to see if e.g. it’s more stable in polar or non-polar environments!
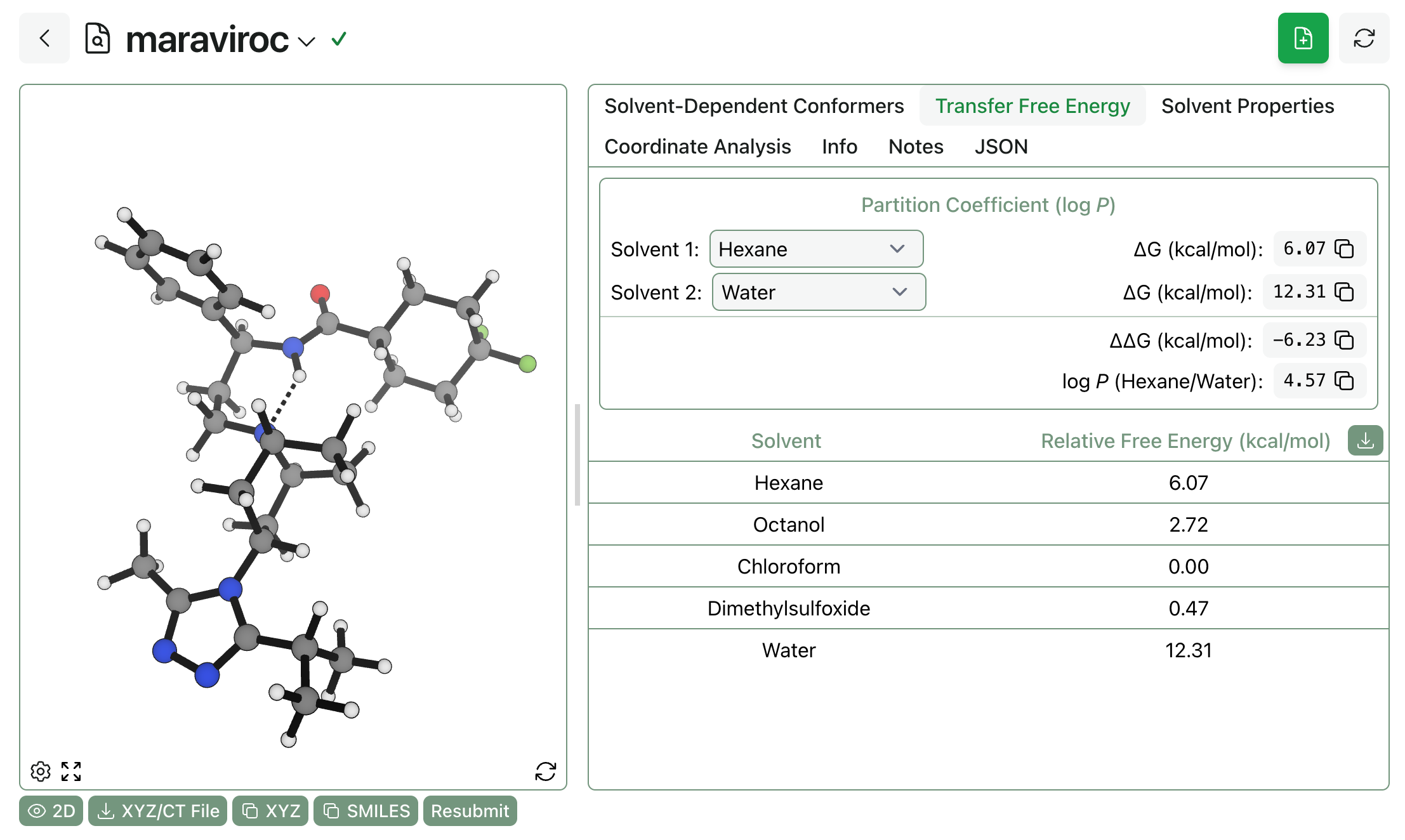
By comparing the overall free energy of the ensembles, you can quickly estimate the transfer free energy between solvents (and thus the partition coefficient). This is similar to the Esol trick that’s proven to be useful for blood–brain-barrier penetrance: but while Esol is limited to just gas–water transfer free energies, this approach allows you to compute transfer energies between any pair of solvents.
Finally, you can also compare Boltzmann-averaged molecular properties between solvents, like radius of gyration or polar surface area. This addresses questions like “how does this molecule’s exposed polarity change between solvents?”, important for designing molecules that can mask polar functionality to diffuse through non-polar membranes.
Our team has been fortunate to have the help and guidance of Rainer Wilcken, one of the original ReSCoSS authors, in building this workflow. Rainer is now a computational chemist at Curie Bio, where he’s worked closely with Rowan to help us develop and validate our low-cost version of ReSCoSS. Here’s what Rainer has to say:
I’m excited to see Rowan develop a lower-cost solvent-dependent conformer workflow. Having a streamlined version of the ReSCoSS concept in Rowan makes it easy for us to roll out across Curie portfolio companies and stay focused on pipeline impact without worrying about managing compute infrastructure.
We’ve been very happy to work with Rainer and Curie in building out this feature, and we’re excited to be able to share this with the rest of our customers.
Running a Solvent-Dependent Conformer Search
Rowan’s solvent-dependent conformer-search workflow is available to subscribing Rowan users. To run a solvent-dependent conformer search, all you need is a molecule (and which solvents to study, if you want to change the defaults):
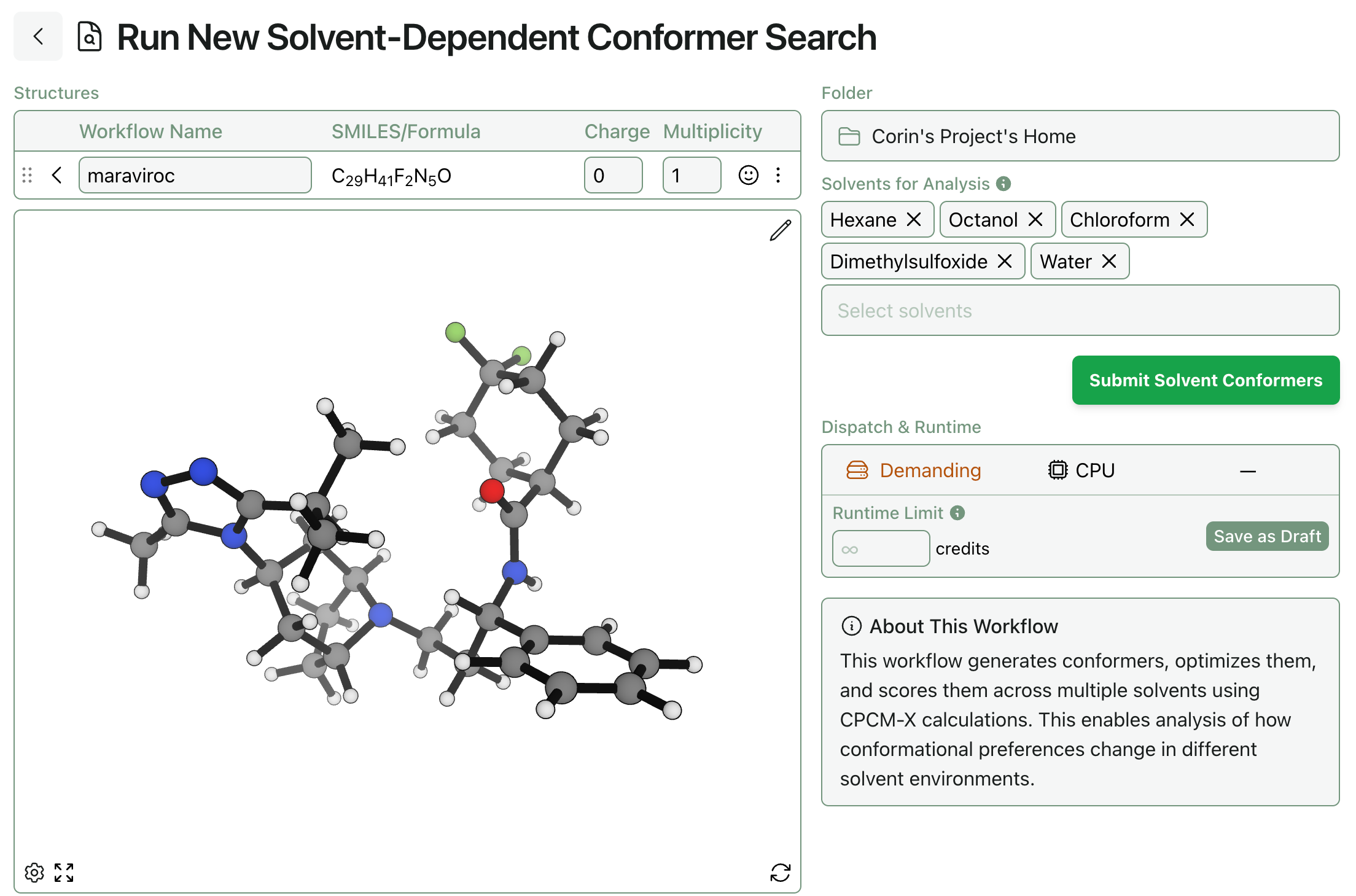
Once the job has finished running, you can see both (1) each conformer’s relative energy and population across all solvents and (2) the ranked list of conformers in each solvent. (You can view this calculation yourself online.)
It’s easy to compute transfer free energies and partition coefficients using the workflow’s output. Here, we can see that the logP(hexane/water) is predicted to be 4.57 for maraviroc (which, admittedly, might be protonated under physiological conditions):
You can also visualize how different internal coordinates vary in different ensembles. Displaying conformer energy as a function of the H55–N4 bond distance shows that internal hydrogen bonding is strongly favored even in water:
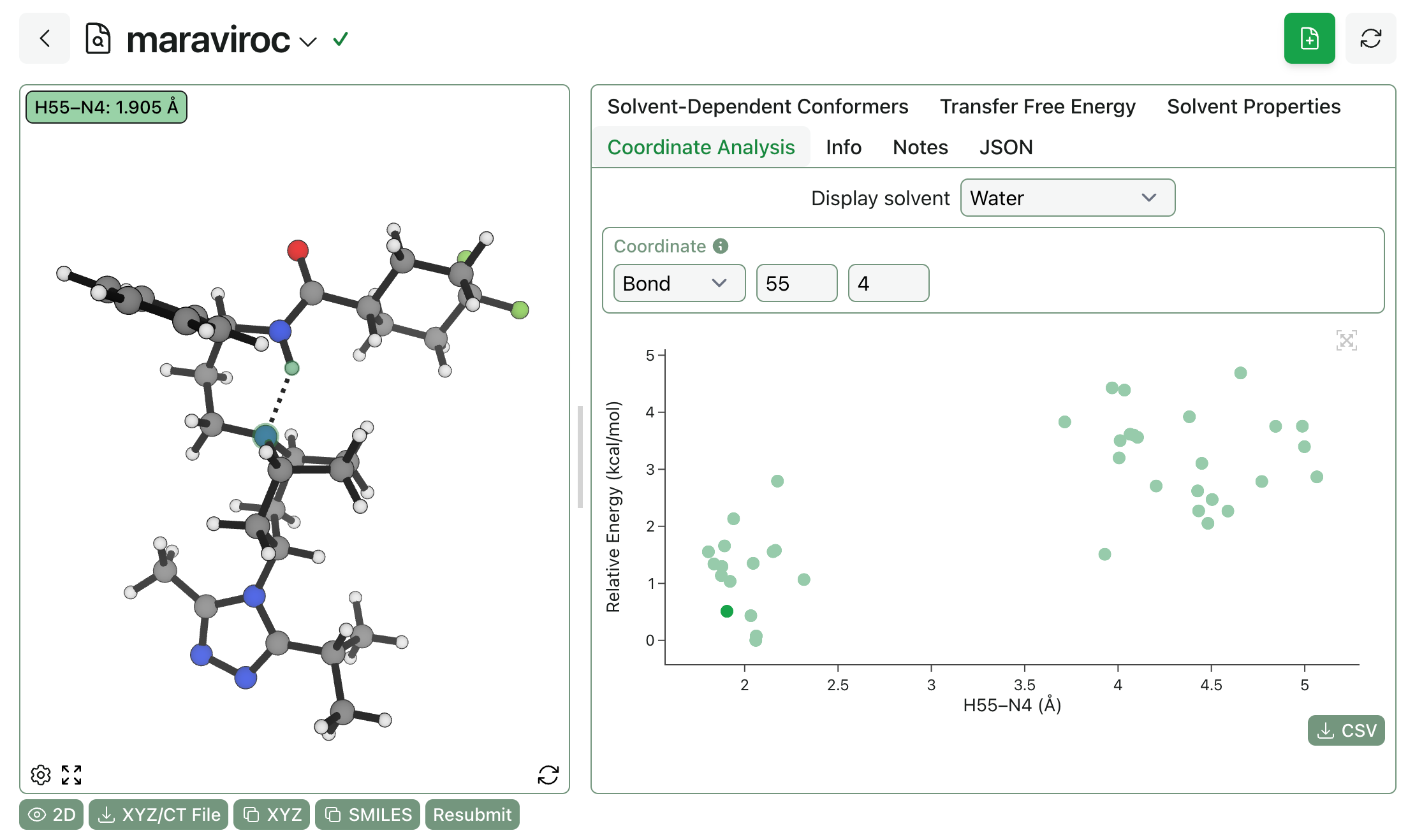
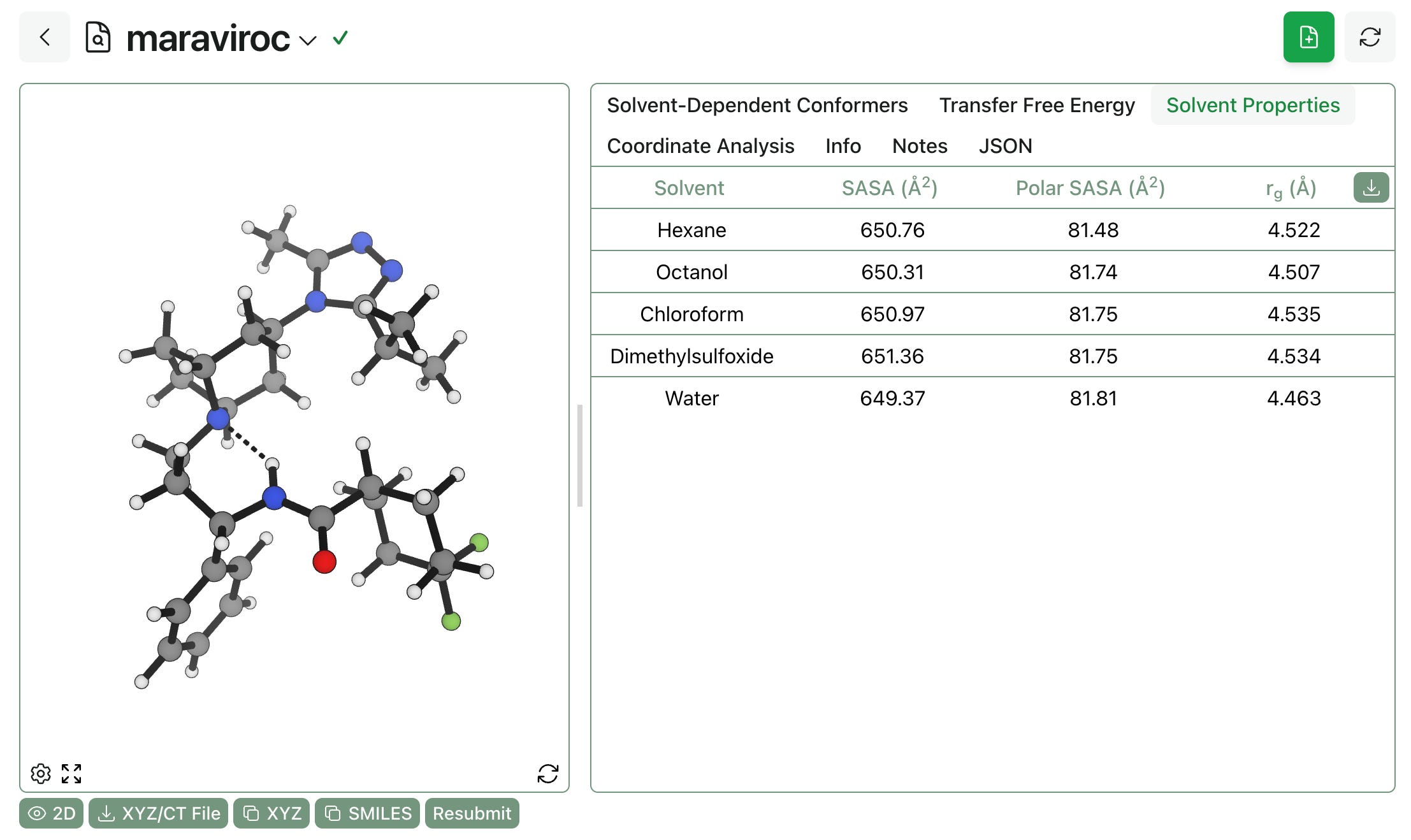
Finally, it’s possible to view the Boltzmann-averaged 3D properties of the entire ensemble. In this case, the strong intramolecular hydrogen bonding observed in all solvents means that maraviroc doesn’t change shape or surface area much across solvents, although it’s worth noting that the aqueous ensemble exposes slightly less total surface area and slightly more polar surface area, as we might expect. (More flexible molecules, like bifunctional degraders, will show bigger differences.)
We’re excited to release this workflow to our customers today. If you’re interested in trying this out, please reach out or subscribe! We’re working hard to bring state-of-the-art drug-discovery tools to all of our therapeutics customers and would be delighted to sit down with your team to understand your scientific challenges & how we can best accelerate your pipeline.