
You Can Now Subscribe to Rowan (And Why You Should)
our vision for Rowan; new pricing model; academic plans; bond-dissociation-energy workflow; improvements to our API




At Rowan, we’re trying to build a fundamentally new form of scientific software.
Existing scientific software packages require years of experience to use properly, and as a result computations are virtually never run by “normal” experimental scientists: instead, dedicated computational experts translate experimental questions into computational workflows, perform the calculations, and then interpret the results back to the experimental scientists. This process is time-consuming and often frustrating; computational scientists complain that experimental scientists don’t take their results seriously, while experimentalists often feel that their theoretical colleagues don’t understand the practicalities involved in their data.
As simulation becomes faster and more powerful, we think it’s crucial that normal scientists are able to benefit from these advances and participate in simulation themselves. Not everyone will become an expert in quantum mechanics or molecular dynamics, of course, but we think that everyone who works with molecules can benefit from the power of modern modeling techniques.
Our job, then, is to create a platform that brings the power of computational chemistry to any scientist. This looks like a lot of things:
Trusted and transparent workflows that encode current best practices for a given task, so you can be sure that you’re getting high-quality results.
A focus on speed, because research moves fast and there’s no point in running a computation that’s slower than an experiment.
Publication-quality visuals so you can immediately understand and interpret your results, even before they’re finished.
One single platform for viewing, submitting, running, and analyzing calculations through lots of different computational backends—because it’s burdensome to ask our users to familiarize themselves with five different point solutions for a single workflow.
And a pricing model that makes it possible to package this all together in a sustainable way.
Ordinarily, the work that Rowan does would be split between several different software packages and platforms, each with their own pricing model:
Scientific software packages are commonly sold with yearly per-seat or per-site licenses, often with additional restrictions on the number of calculations that can be performed. (These prices are often pretty opaque and out of the range of what smaller companies can afford.)
Calculations are run with on-premise computing clusters or cloud computing resources: on-premise computers are purchased outright or rented, while cloud compute is typically billed per usage. (Lightweight calculations can be run on a personal computer, but this quickly becomes burdensome.)
For big companies with internal tools like what Rowan provides, internal developers are required to build and maintain these tools. Plus, the servers, databases, and other computational resources will require their own computers to run on…
To date, we’ve just been charging people for the compute time that they use, akin to how AWS or other cloud providers operate. We’ve grown a little disillusioned with this, though. We want our software to be a trusted partner in your scientific journey, and being acutely conscious that you’ll be billed for each added calculation disincentivizes people from running calculations. (It’s like lawyers: we love our lawyers here at Rowan, but we’re very aware that we’ll get billed for the questions we ask them, which makes us hesitant to reach out!)
Also, charging for usage creates perverse incentives for us as a company. For routine geometry optimizations, I think users should mostly be using AIMNet2 these days, but it’s better for a usage-based pricing model if everyone runs expensive ωB97X-V/def2-TZVPP calculations instead! We want to make our calculations as fast as the laws of physics allow, and we want a business model that aligns our incentives in this direction.
Our New Pricing Model
All this to say—today, we’re launching a paid subscription tier for users with recurring needs. For $200 per month, each user gets 1250 credits per week (a bit over 20 hours of computer time), access to advanced workflows (including our new bond–dissociation energy workflow), and priority support. We anticipate that this will be ideally suited for practicing computational chemists in industry or experimental chemists with frequent computational needs. 20 hours per week should be enough time that you never have to worry about running out of credits—but if you are close to running out, you can configure your usage limit to allow for overages.
We think this is a great deal: for a fraction of the cost of an enterprise software platform, you get access to cutting-edge methods, workflows, hosting, and visualization. There’s no need to renegotiate yearly license contracts or pay for the newest version of a given package, and you can use Rowan for as long as you want or as little as a single month. As we continue to ship new updates that make our software faster and more capable, the value of this subscription will only increase. (This isn’t abstract—we have a lot of updates in the pipeline that we can’t wait to share!)
We’re leaving our free tier more or less unchanged—every new Rowan user will continue to receive 500 credits upon signup. Instead of 20 weekly “low-cost jobs,” we’re giving free tier users 20 weekly credits. This gives users a bit more flexibility in what they can run, and simplifies the experience of using Rowan. (We’re not rolling it out today, but we’ll eventually cap the number of additional credits that free tier users can buy.)
Over the past few months, we’ve massively increased the speed and size of our computing instances to help make difficult DFT calculations faster for big systems, but we haven’t updated our prices to take this into account. Today, we’re also increasing the price of our separately purchased credits from $0.02/credit to $0.05/credit, to match the specs of our new backend infrastructure and ensure we’re not losing money on every calculation.
Although we’re not quite ready to launch it yet, we’re building out a solution for businesses behind the scenes. As a modern web-based computational platform, there’s a lot of things we’re excited to do around collaboration and internal sharing—if you’re interested in being a design partner with us and helping to shape what this looks like, please reach out!

We love our academic users and want to make sure they can keep using Rowan for their projects—accordingly, we’re increasing the amount of weekly credits that we give to academics on the free tier, and lowering the price of the individual subscription to 1/3 of that for industry. (If you’re already an academic user of Rowan, you’ll automatically get the new usage limits today.) If you’re interested in using Rowan for your research group or department, please reach out!

Bond-Dissociation Energy
We’re also launching a bond-dissociation energy workflow today for our paid-tier users. This workflow computes bond strengths, which can be used to predict potential sites of metabolism, site-selectivity in organic reactions, or decomposition pathways for materials. Bond-dissociation energies (BDEs) for simple species can be looked up in reference tables, but for more complex molecules it’s typically necessary to either “guess” BDEs by analogy to another system or compute them from scratch. BDE values can be predicted with good accuracy with quantum chemistry, but computing BDE values for each site quickly becomes burdensome, particularly when combining multiple levels of theory for maximum accuracy.
Rowan’s new workflow makes it simple to accurately compute BDE values for lots of sites in a single molecule: we allow users to manually select bonds or automatically select all C–H or C–X bonds, stream updates to the web as individual bond calculations finish, and display outputs in an intuitive visual format. Similar to our multistage optimization workflow, we combine state-of-the-art quantum chemical methods to give maximum accuracy as fast as possible:
“Reckless” mode runs at the GFN2-xTB // GFN-FF level of theory.
“Rapid” mode runs at the r2SCAN-3c // GFN2-xTB level of theory.
“Careful” mode runs at the ωB97X-3c // r2SCAN-3c level of theory.
“Meticulous” mode runs at the ωB97M-D3BJ/def2-TZVPPD // ωB97X-3c level of theory.
In every case, a linear correction is applied to include zero-point vibrational and enthalpy, which are expensive to compute, but are often just a linear shift. Here’s what the output of a BDE workflow looks like:
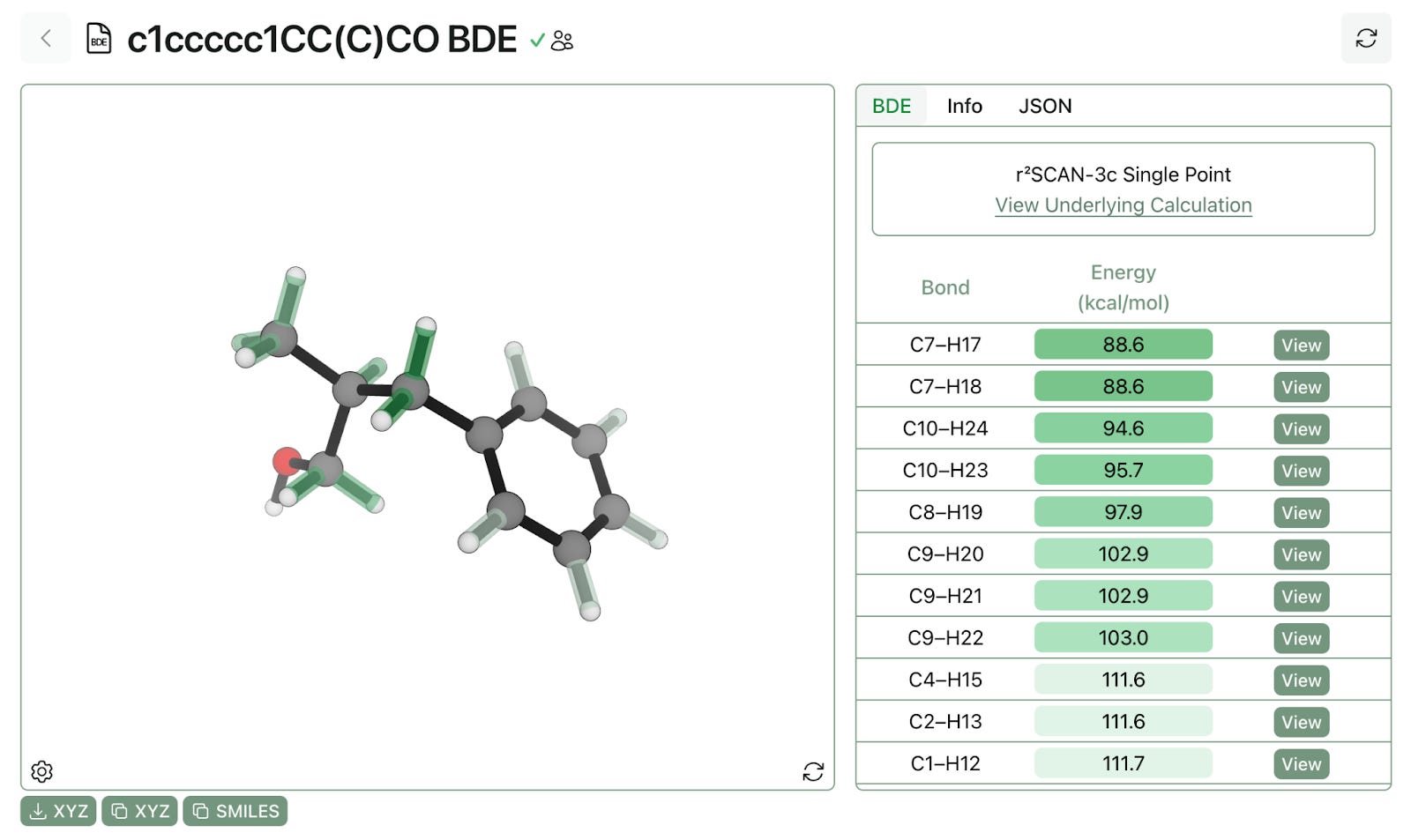
We can quickly see that the aliphatic bonds—and particularly the benzylic bonds—are the weakest bonds in the molecule, and thus the bonds most susceptible to degradation.
Python API
Finally, we’ve completely refactored our Python API. rowan-python 1.0.0 contains a whole new set of methods that make it possible for end users to create, read, update, and delete folders and calculations, while maintaining the existing high-level interface that allows users to automatically submit calculations and wait for the result. This can be very simple:
import rowan
folder = rowan.Folder.create(
name="test Rowan API folder"
)
try:
result = rowan.compute(
workflow_type="basic_calculation",
molecule="CC(=O)OC",
name="test",
settings={"method": "gfn2_xtb", "tasks": ["energy"]},
engine="xtb"
)
print(result)
finally:
rowan.Folder.delete(folder["uuid"])We’re really excited about the potential for Rowan to serve as the high-level backend for a variety of quantum chemistry applications: if you’re working on something in this area, please reach out!