
Boltz-2 Constraints, Implicit Solvent for NNPs, and More
new terms of service; comparing IRCs and conformer searches; contact and pocket constraints for Boltz-2; MOL2 download; implicit-solvent NNPs; draft workflows; optimizing docking efficiency




We’re excited to share a variety of quality-of-life improvements across the Rowan platform.
New Terms of Service & Purchased Credit Expirations
We've updated our Terms of Service, effective September 17, 2025. The terms now include information about weekly and purchased credits, annual subscriptions, and subscription-renewal notices.
Every time a calculation runs on Rowan, it uses credits. One credit equals one minute of CPU/GPU time. We offer two types of credits: weekly credits and purchased credits. Weekly credits are included in subscriptions and refresh each Sunday. Purchased credits are additional credits you buy whenever you need more. As of Wednesday, purchased credits remain in your account for up to one year from the purchase/issue date. All credit purchases are final and non-refundable. Credits purchased before September 17, 2025 will expire on September 17, 2026.
We announced our new annual plans (with discounted pricing!) three weeks ago. If you’re subscribed to an annual plan, we'll send you a renewal notice 30 days before your plan renews.
If you’re a Rowan user, please take a moment to review the updated Terms of Service (if you haven’t already).
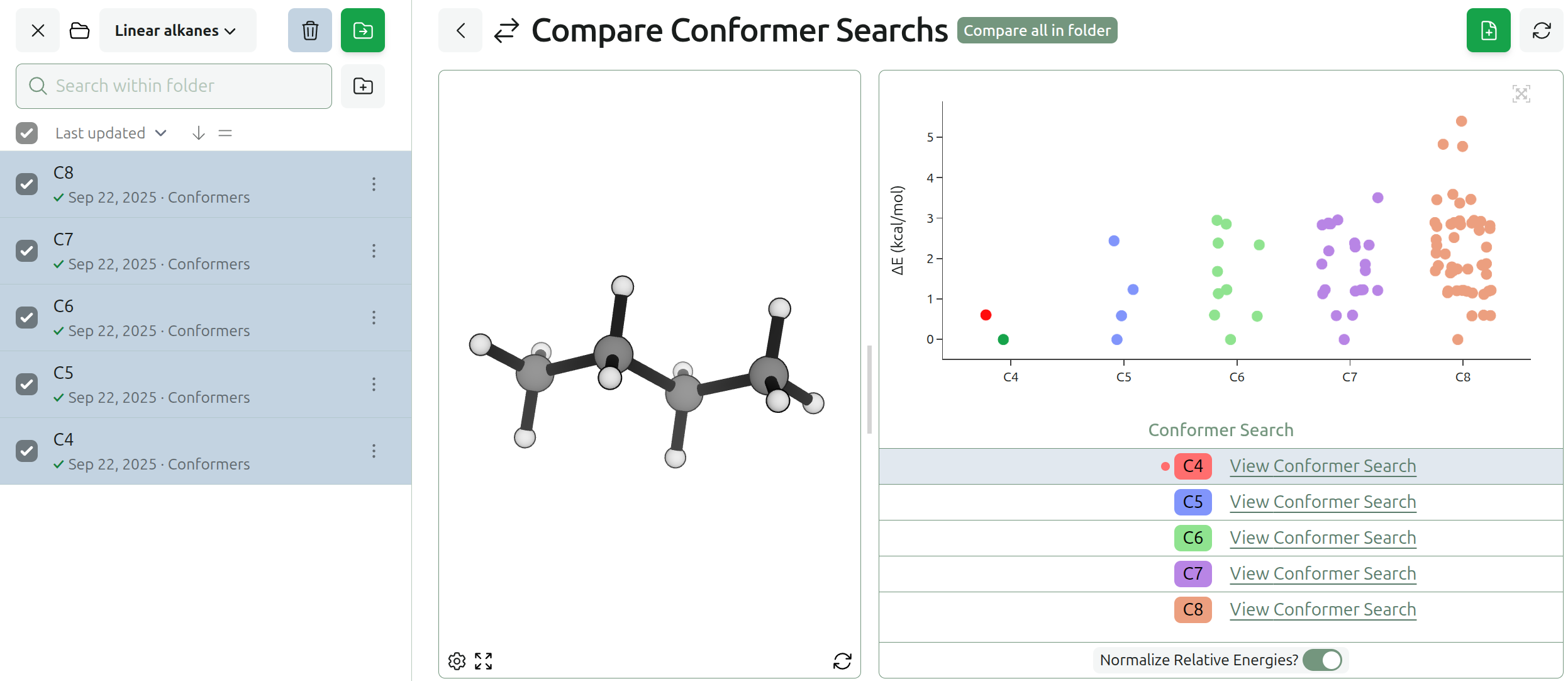
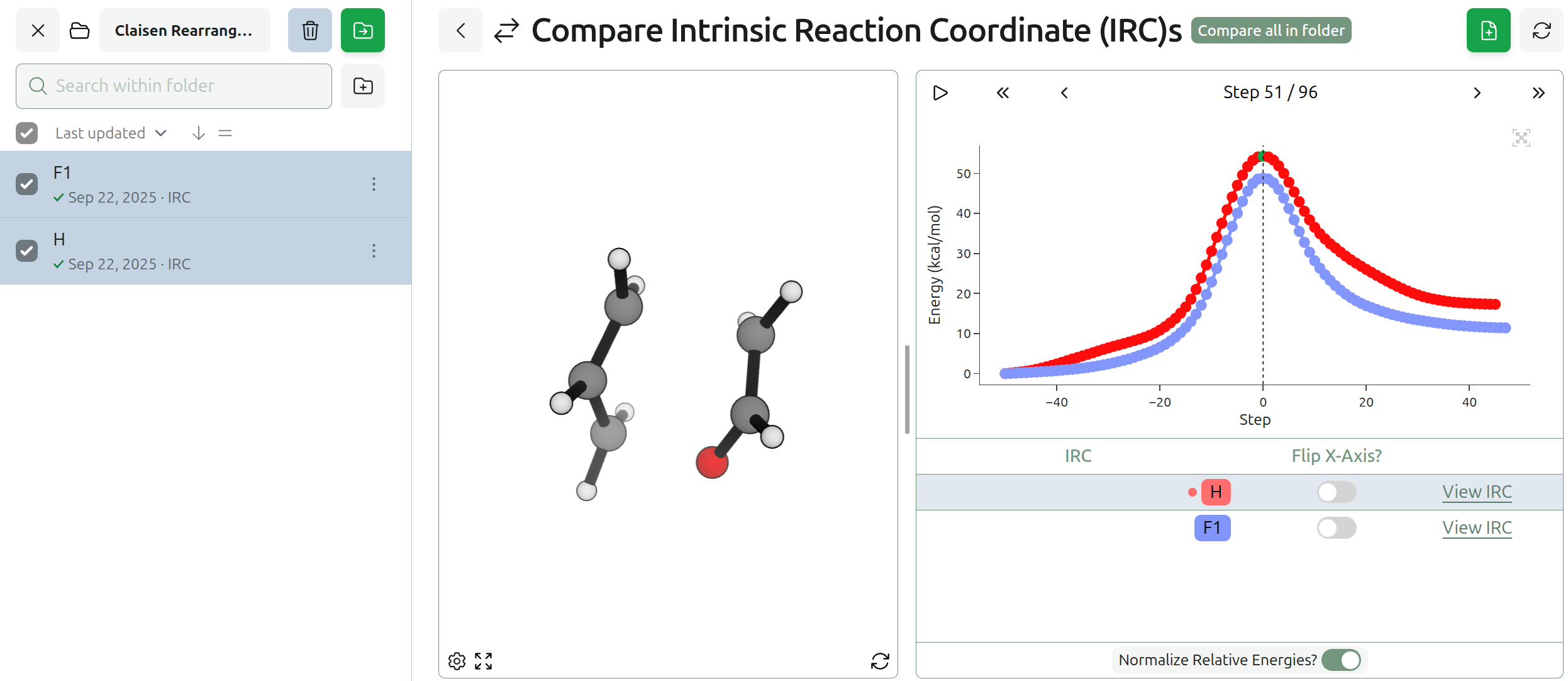
New Compare Views for IRCs & Conformer Searches
We’ve also added the ability to compare conformer searches and IRCs.
Various conformer search methods can produce different numbers and distributions of conformers, and small changes to a molecule can produce significant changes in the preferred lowest-energy conformer. For the linear alkane series, the number of low-lying conformers quickly increases with each additional carbon. However, the lowest-energy conformer remains the unbent version until ≈16 carbons (see work from Byrd and co-workers).
One can also compare intrinsic reaction coordinate (IRC) calculations, which makes it easy to assess how the reaction path differs between various substrates or levels of theory.
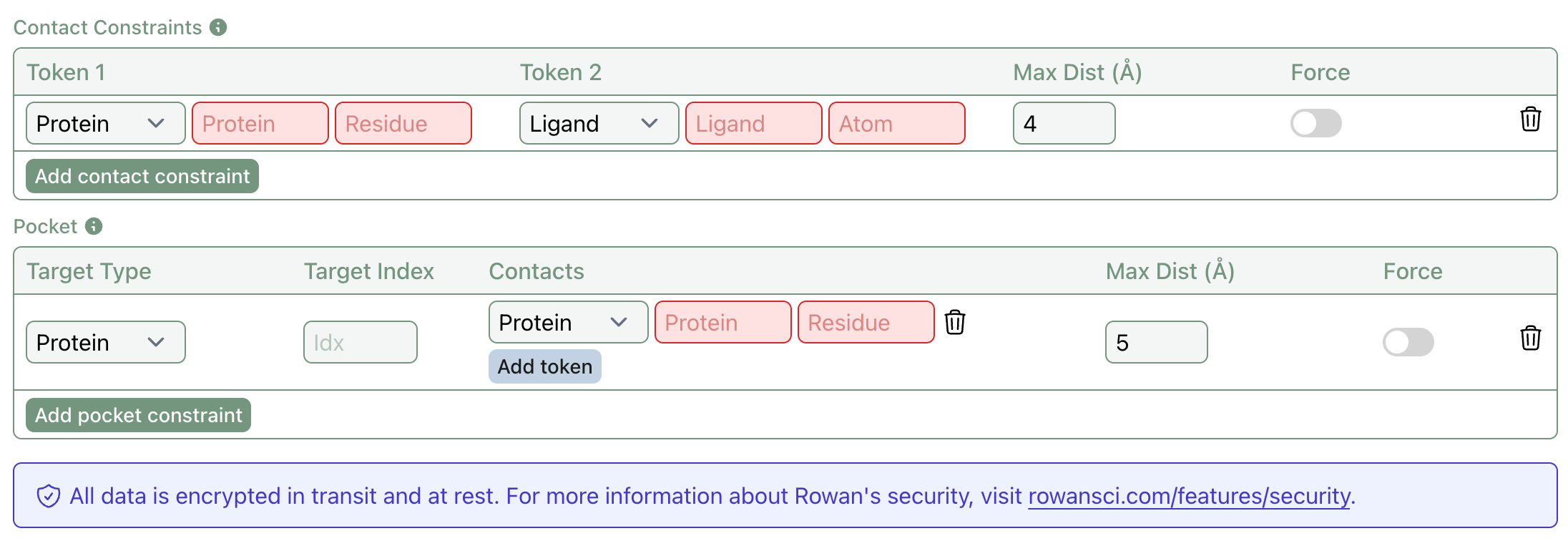
Boltz-2 Contact & Pocket Constraints
We’ve added support for two types of constraints to Boltz-2: contacts and pockets.
To briefly explain how these two types of constraints work, it's helpful to remember that AlphaFold3-style models process inputs (protein chains & ligands) as a series of tokens. For protein chains, each residue is one token. For ligands, each atom is one token.
Contact constraints let users specify that two tokens, such as a ligand atom and a protein residue, should remain within a defined distance. This helps bias co-folding predictions toward known or suspected interactions.
Pocket constraints are coarser, letting users specify that a token (or multiple tokens) and an input (a protein chain or ligand) should remain within a defined distance. This could be used to dock ligands into a known binding site by specifying that a ligand should be close to a number of residues forming a known pocket. (For more details about these constraints, see the Boltz documentation.)
Both features support optional potential forcing, giving you the choice between gently guiding and strictly constraining the co-folding process. Together, these tools make co-folding predictions more targeted, interpretable, and biologically relevant.
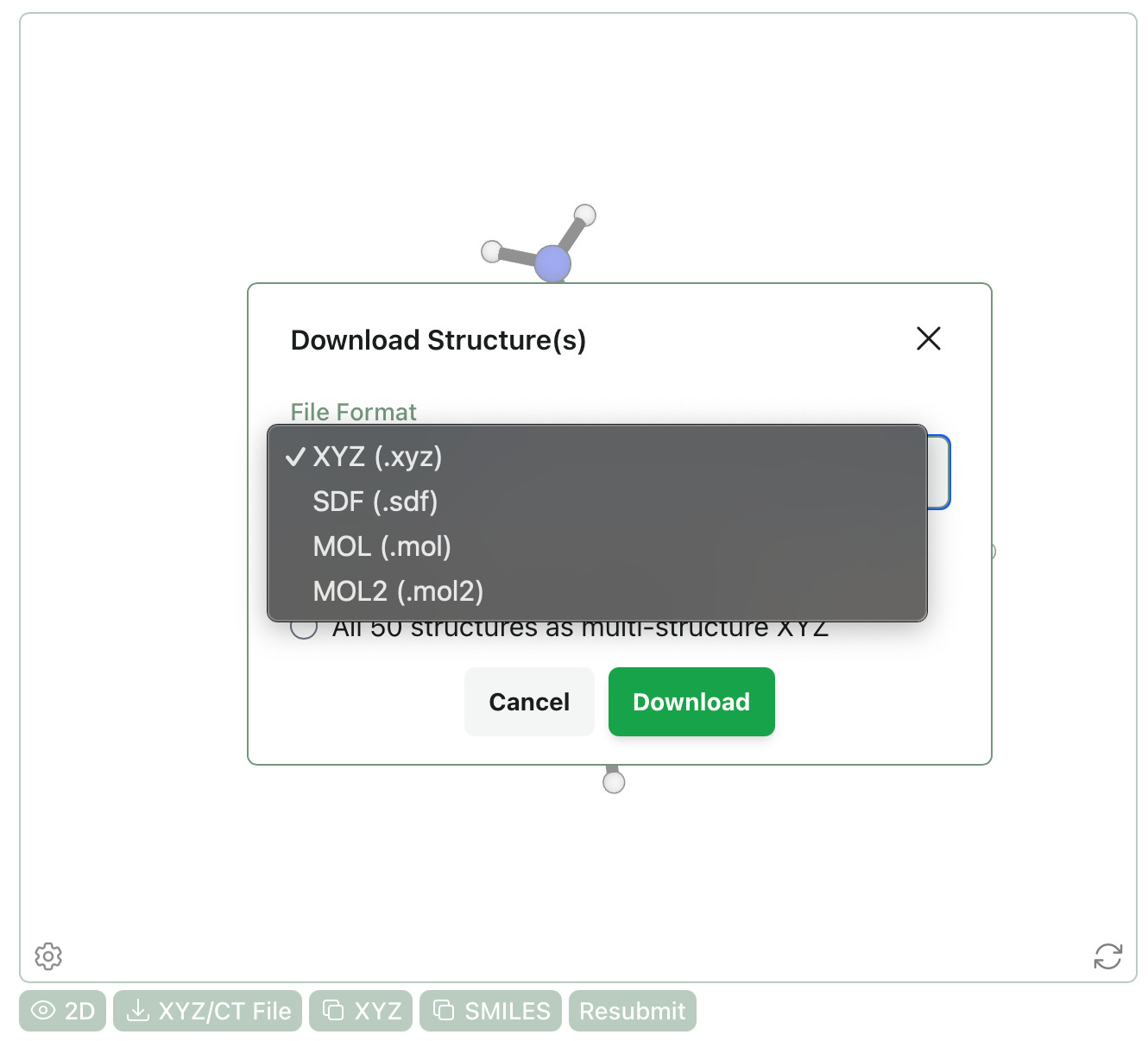
Download MOL & MOL2 Files
We’ve added the ability to download molecular structures as MOL and MOL2 files. Unlike XYZ and SDF, which support multi-structure files, MOL and MOL2 are limited to single-structure files. If other formats would be useful for your work, please let us know at contact@rowansci.com!
ALPB Solvent Model for NNPs
Neural network potentials are approaching DFT accuracy for many gas-phase modeling tasks, but most NNPs don’t allow users to specify an implicit-solvent model, which limits their applicability to many chemical problems. Previously, we’ve used semiempirical implicit-solvent calculations to correct NNPs, like in our microscopic-pKa-prediction workflow and our recent redox-potential study—now we’ve added ALPB solvent corrections to all NNPs, making it possible to incorporate solvent effects alongside UMA, eSEN-OMol25, Egret-1, AIMNet2, and other models.
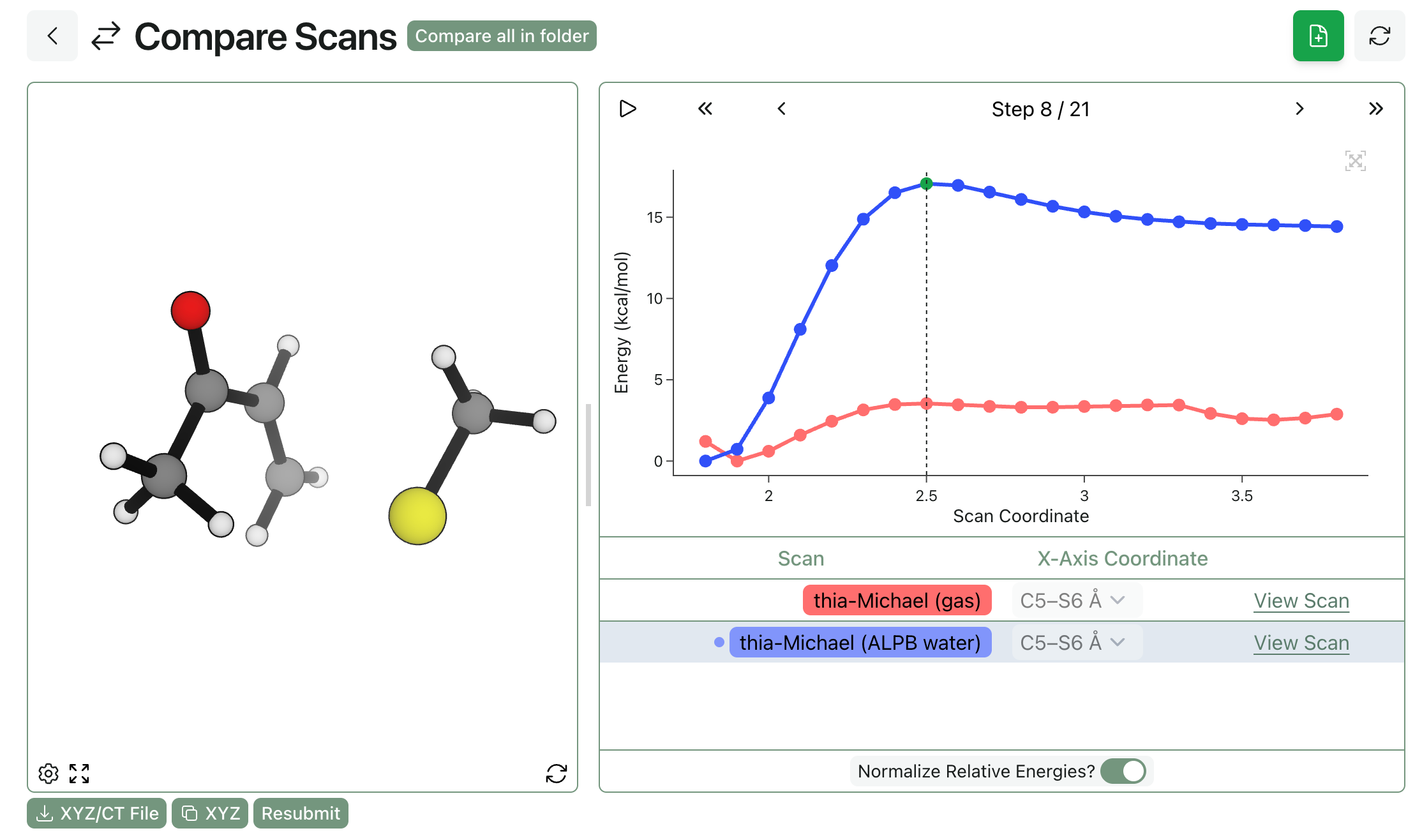
This can make a massive difference. We studied the below thia-Michael reaction at the eSEN-OMol25-sm-conserving level of theory, both with and without implicit water. The scan run with implicit water (shown in blue) has a clear product well and maximum; in contrast, the gas-phase scan (shown in red) has no clear barrier and is pretty noisy overall.
Draft Workflows
After some helpful feedback from our users, we’ve added the ability to save workflows as drafts and resubmit them later. This makes it simple to save structures that are partially edited without copying them to an external file.
Create
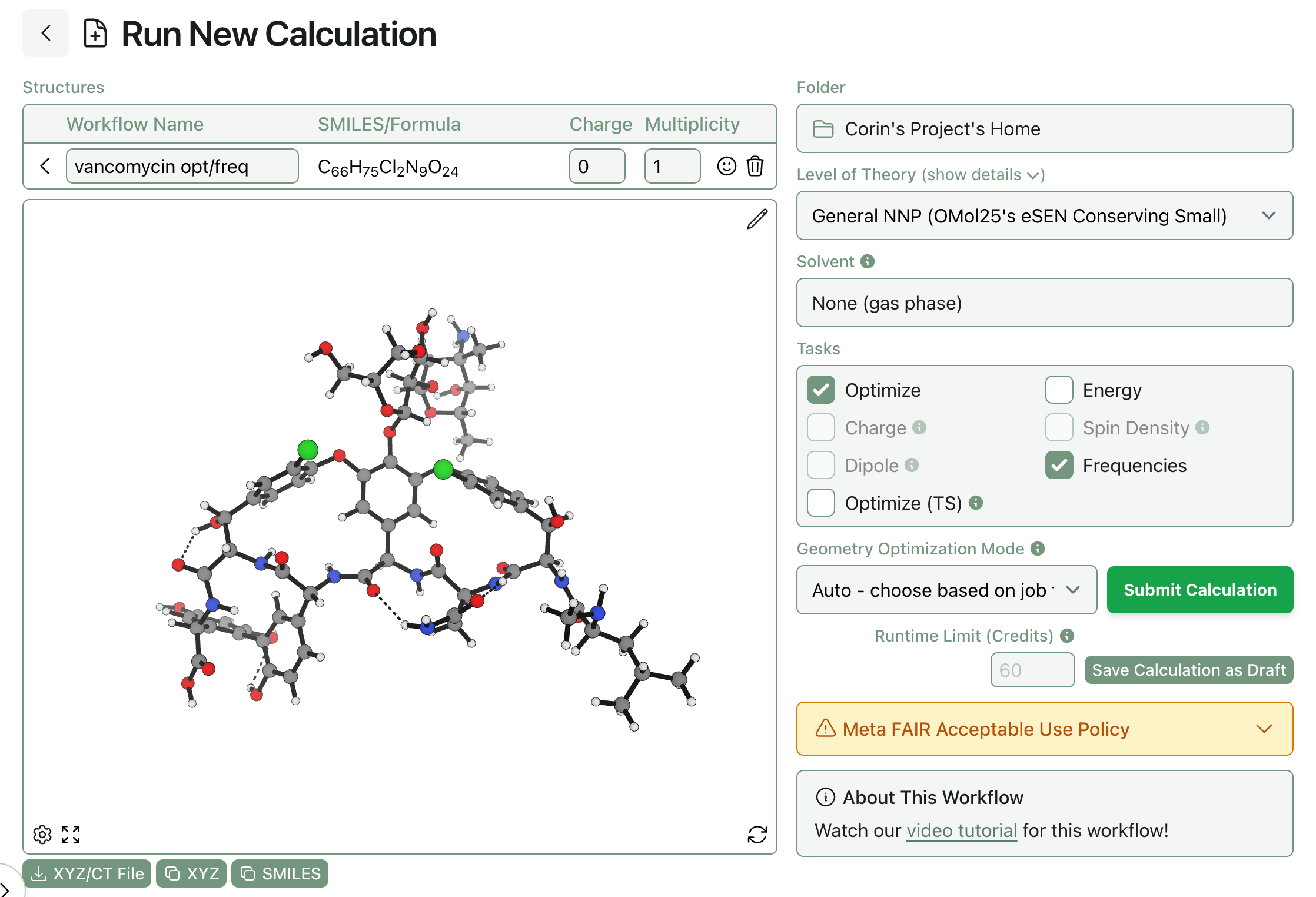
Draft workflows can be created by clicking “Save Calculation as Draft” instead of “Submit Calculation” on the workflow submission page.
Edit
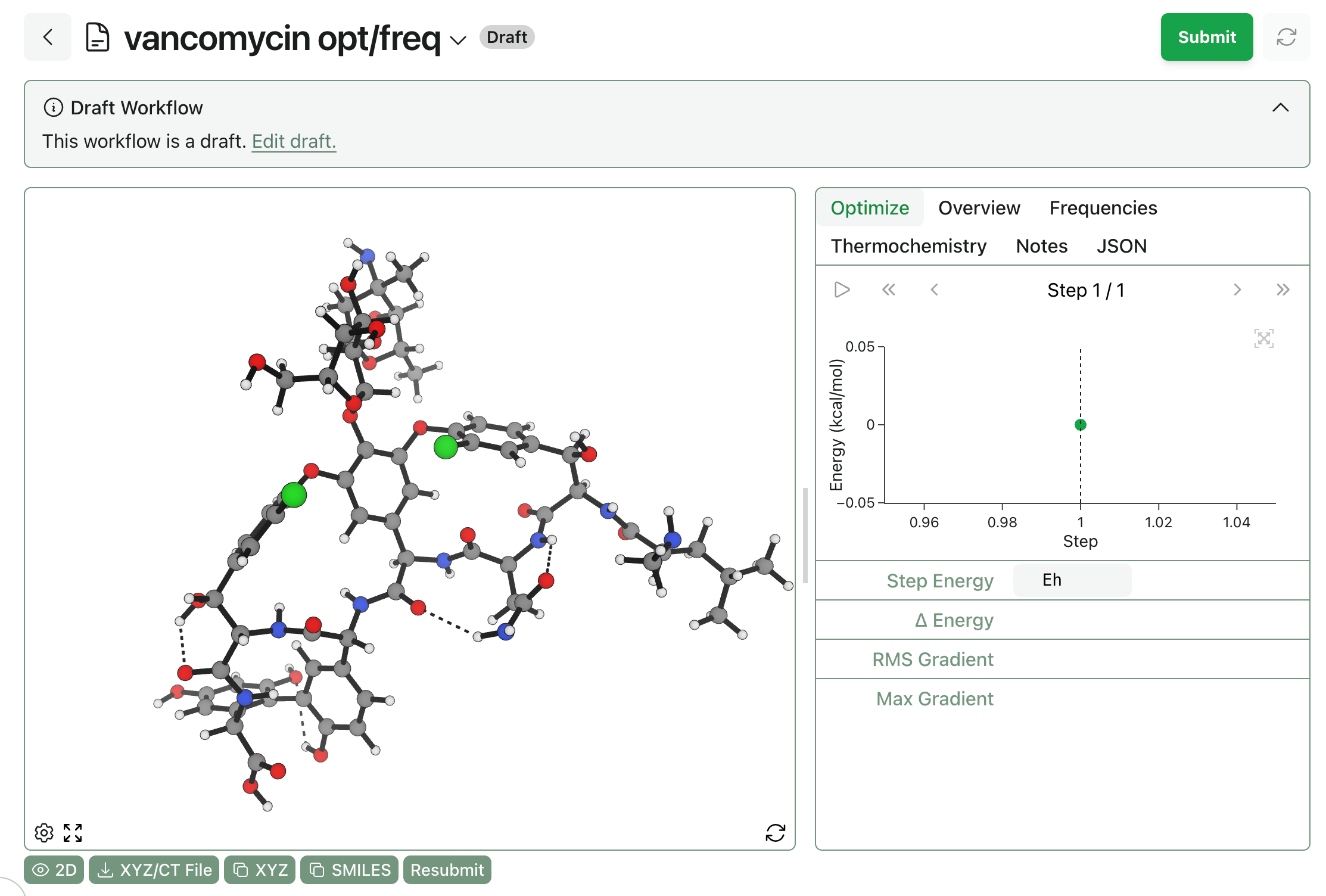
Drafts can be viewed just like any other workflows. To edit a draft, click “Edit Draft” when viewing the workflow—this allows users to change the type of workflow, edit the structure, and change any workflow-specific settings.
Submit
A draft workflow can be submitted by clicking “Submit” when viewing the draft workflow. This will move the workflow into the “Queued” or “Awaiting Queue” states, depending on the number of currently running jobs.
How Long Does Docking Take?
As more users begin to build on Rowan’s Python API, we’ve started to see people run lots of docking calculations in parallel. To help scientists run the most efficient virtual screens, we recently ran a study comparing how different docking settings impacted the accuracy and runtime of the resultant workflow. We recommend (1) setting do_pose_refinement to False if the geometry of output poses doesn’t matter and (2) disabling the conformer search step for efficient high-throughput screens: read the full post here.