
Structure-Based Drug Design Updates
enforcing stereochemistry; refining co-folding poses; running PoseBusters everywhere; computing strain for co-folding; PDB sequence input; 3D visualization of 2D scans



Over the past year, we’ve added a number of structure-based drug design (SBDD) tools to Rowan: we launched docking back in March, protein–ligand co-folding with Boltz-1 and Chai-1 in May, and co-folding with Boltz-2 in June. While these tools are used throughout the drug-discovery industry, they’re flawed. Neither docking nor co-folding can consistently predict relatively binding affinities on unseen targets, and naïvely trusting the output of either workflow without additional checks is likely to lead to disappointment.
Since we launched these tools, we’ve talked with a lot of excellent scientists working in early-stage drug discovery about how they use docking and co-folding in the real world. We’ve learned a lot from these conversations. Today, we’re releasing a suite of new features aimed at implementing these current best practices into Rowan.
Our goal is to make it easy for anyone using Rowan to use these workflows the same way that scientists at top-20 pharma companies or leading biotech startups use them—with lots of safeguards and checks in place to ensure that the results are as useful as possible. Here’s how we’re modifying our structure-based drug discovery workflows to make them more robust.
Automatic Stereochemistry Checks
During inference, Boltz-2 and other co-folding models generate the 3D structure of a ligand from an input (typically a SMILES string). Unfortunately these models often generate these ligands with incorrect stereochemistry: Boltz-2 is known to flip stereocenters and generate incorrect enantiomers or diastereomers for small-molecule ligands. These issues are documented on GitHub and Rowan users have reported this error as well.
While we’re not able to fix the underlying scientific problems here, Rowan’s co-folding workflow now automatically checks the stereochemistry of the output conformation and alerts the user if this stereochemistry is wrong.

Pose Refinement For Co-Folding
Most co-folding algorithms ignore hydrogens and output molecules with questionable geometries. Rowan now automatically adds hydrogens and, with “Refine Pose?” selected, runs constrained local optimizations to “clean up” geometries, allowing downstream applications like molecular dynamics to run without crashing (and making it a lot easier for chemists to view the output poses).
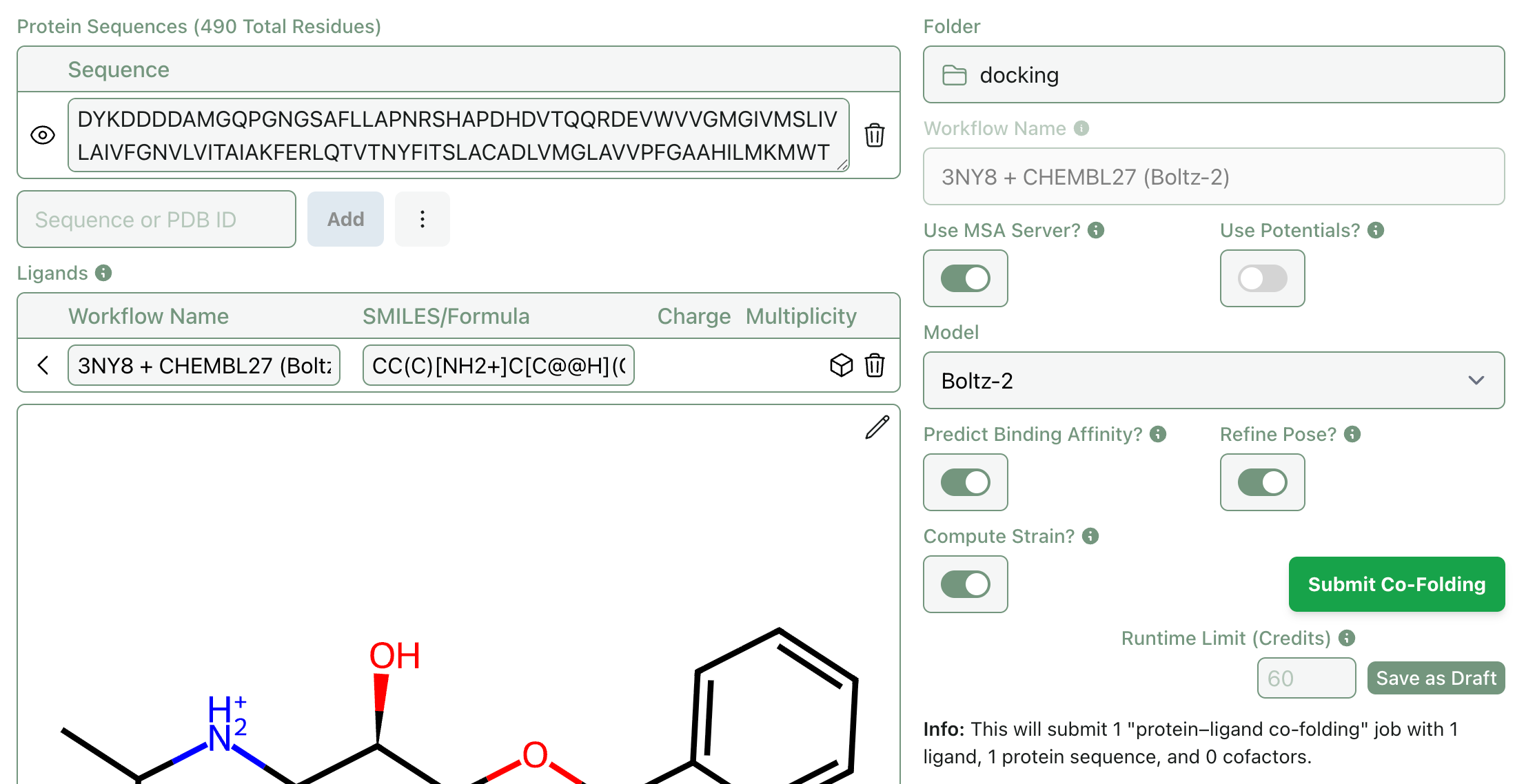
Both the refined and original PDB files can be downloaded.
Automatic PoseBusters Analysis & Filtering
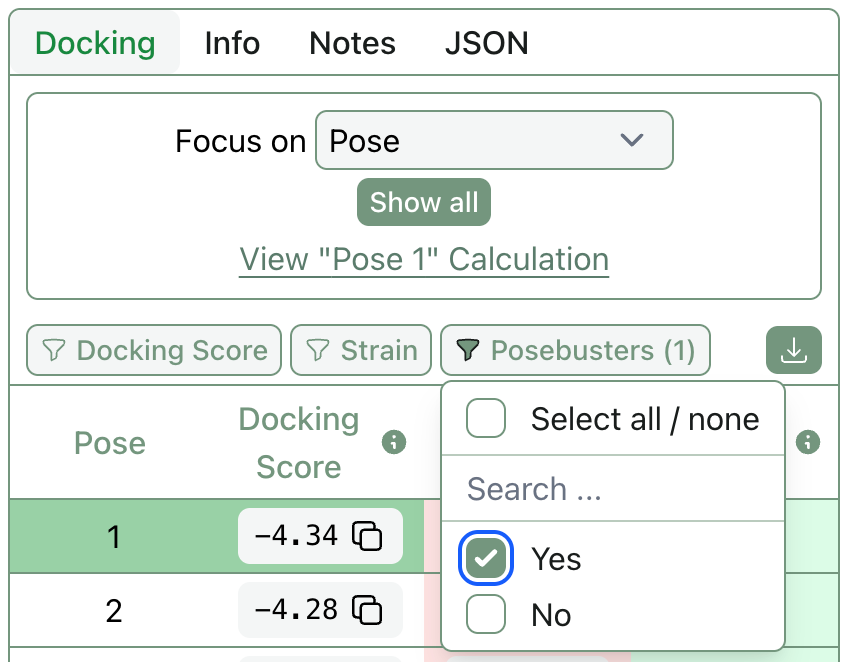
In 2024, Charlotte Deane and co-workers reported that most AI-based pose-generation methods generated physically implausible conformers. While more recent algorithms have taken strides towards fixing these issues, it’s still not uncommon for a co-folding algorithm to return wacky poses. Rowan now runs the recommended “PoseBusters” suite of structural checks against all docking and co-folding poses, allowing users to quickly assess whether a given pose is physically reasonable or not.
It’s also easy to filter out all docked poses that failed PoseBusters with our new pose-level filtering.
Co-Folding Strain Analysis
Since launch, Rowan has automatically used AIMNet2 to compute the strain energy of docking-generated poses, allowing users to filter out unreasonable poses and understand the energetic penalty associated with various conformations.
We’ve now added this feature to our co-folding workflow, as shown at the bottom of the below screenshot. As with docking, excessively high strain energy indicates that a generated pose is energetically inaccessible and probably unphysical.
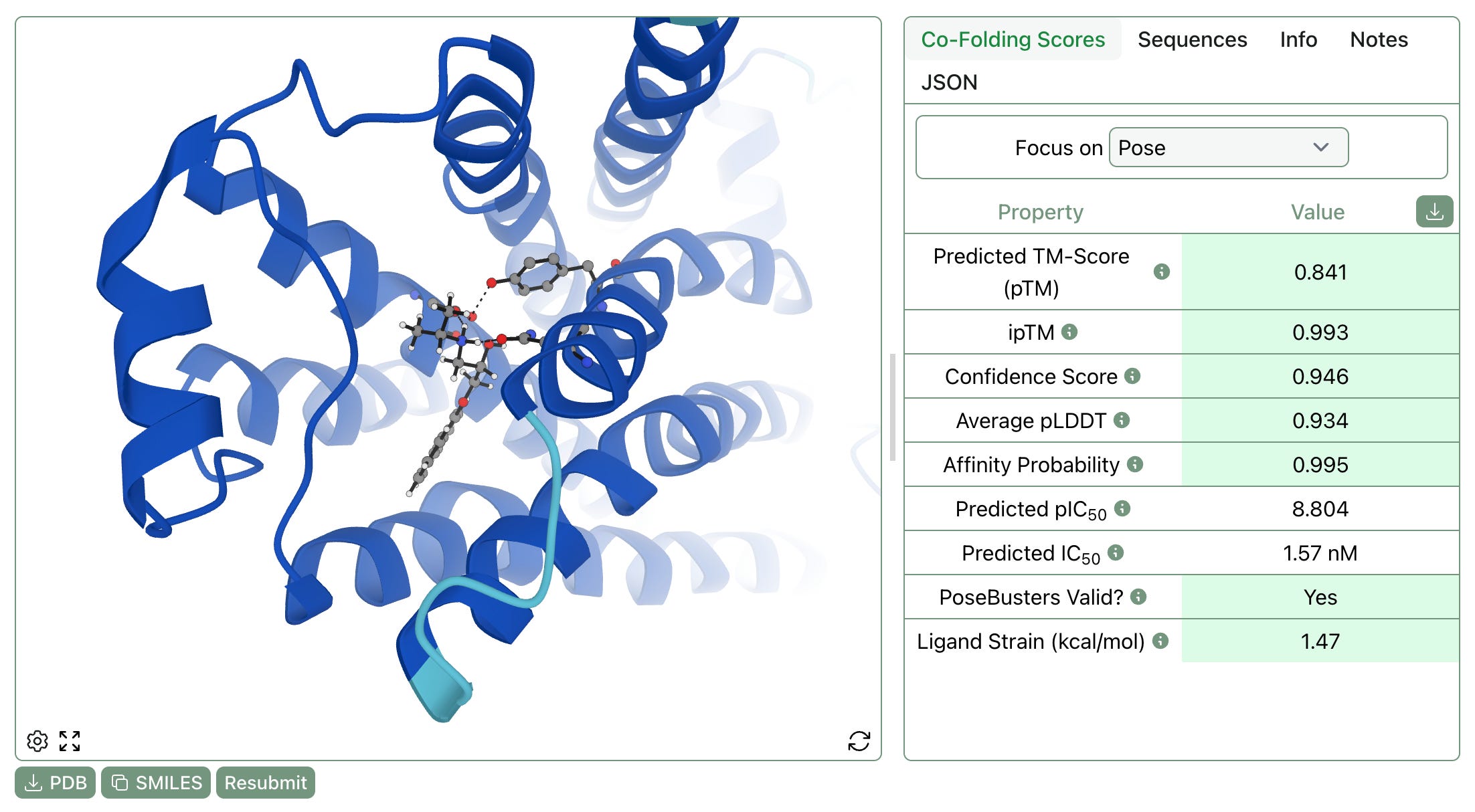
Strain calculations can be enabled by selecting “Compute Strain” on the workflow submission screen.
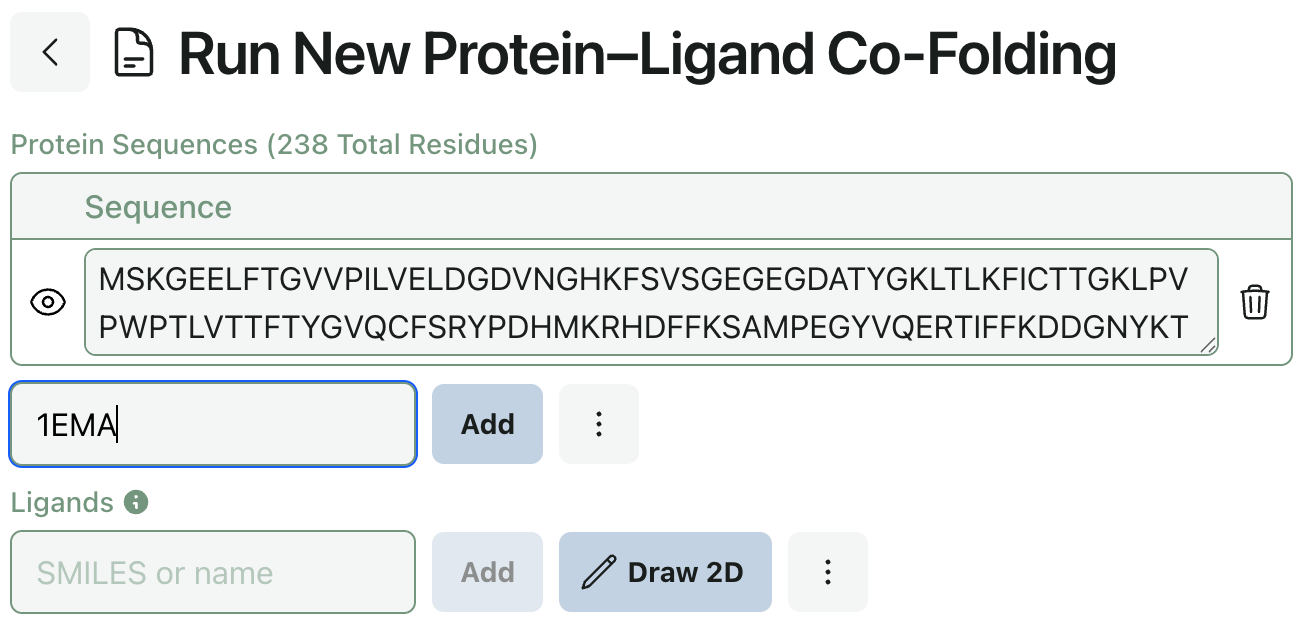
Input Sequences from PDB ID
In response to user feedback, we’ve added the ability to input protein sequences via PDB ID. This makes submitting co-folding jobs with known proteins much simpler (and keeps you from having to switch tabs).
3D Graphs for 2D Scans
Finally (unrelated to SBBD), we’ve added a 3D graph view to 2D scan outputs. While not technically super useful or new, we think this is pretty cool (and users asked for it). Here’s what this looks like for a Diels–Alder reaction:
Until next time, happy computing!